鋰離子電池漿料的制備技術及其影響因素
2021-07-08 09:10:26歐陽麗霞武兆輝王建濤
材料工程 2021年7期
歐陽麗霞,武兆輝,王建濤
(1 有研科技集團有限公司 國家動力電池創新中心,北京 100088;2 國聯汽車動力電池研究院有限責任公司,北京 100088;3 北京有色金屬研究總院,北京 100088)
鋰離子電池作為一種新型的高性能可充電電池,因其獨特的優點而得到廣泛的研究和應用,主要包括正極、負極、電解液和隔膜。其工作機理是鋰離子通過電解液往返于正負電極間嵌入和脫出的過程,故正負極電極的性能對于鋰離子電池總體性能起決定性作用[1]。
制造鋰離子電池電極最常用的工業技術為濕法處理。盡管已被廣泛接受,但該處理方法仍面臨著許多的問題,包括昂貴且危險溶劑的回收、涂層的不一致性以及由溶劑干燥過程所導致的微觀結構的變化[2]。這些問題都與制備鋰離子電池的第一道工藝——漿料的性能有著重要關系。
鋰離子電池電極漿料通常包括活性物質、導電劑、黏結劑和溶劑。導電劑為電子傳輸提供通道,黏結劑把活性物質與導電劑黏附在一起并將其黏附在集流器上[3-4]。漿料制備過程為將其組分混合均勻的過程,漿料的性能決定了后續鋰離子電池的性能。漿料體系中不同顆粒間的物理性質如尺寸、形貌不同,顆粒間往往會發生分散或團聚[5-9],造成漿料內部均勻性降低,這會使得鋰電池壽命減小甚至產生安全隱患。
漿料的性能還對電極的生產率有著重要的影響。除去材料之外,電池成本中最昂貴的便是電極制造。電極生產包括漿料的制備、涂布與干燥。而與涂布和干燥相比,得到均勻的漿料所需的時間是限制電極產量的關鍵因素。另外,漿料的質量還會影響其涂布的可加工性。常用的涂布設備除了有工業要求的操作速度,還要能精確地實現均勻的涂布厚度。但通常的涂層仍會存在一些缺陷,如附聚物、縮孔、金屬顆粒污染或組分過度的不均勻,從而影響循環過程中的庫侖效率或倍率性能[2]。而這些缺陷均可以由混合不充分、漿料脫氣不充分或硬件故障產生。
根據研究,漿料的均勻性和穩定性的主要因素有原料組分、溫度、pH值和混合順序等。本文總結了影響鋰離子電池電極漿料穩定性和均勻性的因素,對研制出更加均勻和穩定的漿料體系、制備優質電極和提高電池性能具有一定的指導意義。
1 電極漿料研究遇到的問題
鋰離子電池漿料是一種處于非平衡態的懸浮液體系。為滿足各行業對高性能鋰離子電池的需求,固體顆粒粒徑通常很小,朝著納米方向發展,從而導致顆粒極其容易團聚,以及漿料中各組分的分布不均勻[10-11],影響電極涂層內部的微觀結構和電池的倍率性能,進而對電池的安全性和耐用性均會產生影響。在活性材料、導電劑以及集流體之間存在著導電的網絡連接,漿料內部材料分散越均勻,這種網絡連接的導電性也就越強,從而鋰電池的性能也就越優異。因此,漿料內部物質的分散均勻性對于鋰離子電池的性能有著非常重要的作用[12-13]。
隨著勻漿結束,攪拌停止,漿料會出現沉降、絮凝聚等現象,這對后續的涂布等工序造成較大的影響。因而漿料在制造好之后,在涂敷之前的存儲期間內必須要能夠有一定的穩定性。通常,穩定性定義為漿料承受質量或相分離的能力,允許在混合步驟后長時間保持均勻的顆粒分布[14]。
在實際情況中,漿料本身大多都是深色,用肉眼無法直接觀測內部顆粒分布是否均勻,而根據其為黏性流體或膠體特性,不同的分散狀態對應著不同的流變性,因此,研究者都是通過漿料的流變性能來分析漿料內部均勻性[15-16]。大量研究表明,漿料的分散均勻性和沉降穩定性與原料添加順序、溶劑種類、固含量、攪拌工藝等密切相關[16-22]。
2 漿料分散性和均勻性的影響因素
黏度是流體黏滯性的一種度量,是流體流動力對其內部摩擦現象的一種表示。漿料的黏度通常隨剪切速率而變化,該現象可對漿料中的顆粒-聚合物之間的相互作用進行詳細的描述。當存在剪切變稀行為時,漿料中存在容易被剪切應力破壞的軟團聚物。相反,剪切增稠的存在通常表明漿料中有著硬聚集顆粒。黏彈性也是流變學重要的參考參數。通過儲能模量(G′)和損耗模量(G″)的相對值來表征漿料的黏彈性特征。儲能模量G′又稱為彈性模量,代表漿料發生可逆彈性形變時所儲存的能力,是漿料彈性變形的度量。損耗模量G″又稱為黏性模量,代表漿料發生不可逆變形時消耗的能量,是漿料黏性變形的度量。在頻率掃描中,基于G′和G″的相對大小,并評估G′對角頻率的靈敏度,能夠反映出漿料是流體狀態還是類固體狀態的信息。并且在低頻范圍下,G′>G″且其差值越大,表明漿料的穩定性越好。較為少見的是幅度掃描測試。在幅度掃描中的低應變下,G′>G″且G′的值保持相對恒定,這表明漿料內部存在著網絡結構,且該網絡結構是完整的。該凝聚網絡區域成為線性黏彈性區域,并且G′的常數值被稱為平衡存儲模量(G′0),描述網絡結構的強度。另一種流變性質是屈服應力(σ0),表示誘導漿料流動所需的最小力。
研究顆粒-聚合物體系的內部結構和分散狀態的廣泛方法為流變學。如果顆粒之間存在牢固的結合,彼此之間沒有很好的分散,通過流變學特性發現固體或凝膠狀行為,且其黏度很高。相反,當漿料充分分散時,將出現類似流體的行為,并且黏度將大大降低。
2.1 活性物質對漿料流變性的影響
活性材料的表面狀態會影響著漿料特性和分散狀態。Tsai等[23]研究了兩種表面狀態的磷酸鐵鋰(LiFePO4, LFP)對水系漿料的流體狀態的影響,其中凝膠態磷酸鐵鋰(G-LFP)是對分散態磷酸鐵鋰(D-LFP)進行碳包覆處理后的材料。G-LFP顆粒的表面包覆碳上存在著眾多的碳衍生物,如羧基、羥基和羰基等有機官能團,這些官能團間的相互作用導致G-LFP顆粒在水基漿料中的凝膠化。由圖1(a)中可知,D-LFP漿料為流體特性,但其G′>G″表明懸浮液應該更偏向于凝膠特性[23-24]。這可能由D-LFP漿料具有極高的固體負載量,未分散開的D-LFP顆粒形成的團聚體所造成。對于G-LFP顆粒制備的漿料,其G′>G″表明漿料內G-LFP顆粒也存在著大量的團聚。但G-LFP漿料的G′遠大于G″表明顆粒的凝膠化占主導效應。和D-LFP漿料相比,G-LFP漿料的G′對掃描頻率的依賴性更低,其較高彈性與漿料內部由有機官能團形成的3D凝膠狀結構的較高彈性相關。此外,G-LFP漿料的G′和G″數值均比D-LFP漿料的G′和G″高幾倍。這一實驗結果表明G-LFP漿料中的顆粒之間和顆粒與溶劑之間的相互作用更強。從圖1(b)中可以看出在氮氣(N2)中經過750 ℃處理后的G-LFPΔN2的黏度值遠遠小于G-LFP漿料。這是因為G-LFP經過熱處理之后,其表面的碳衍生物的減少有利于LFP顆粒的解凝膠,使得G-LFP漿料由凝膠狀態轉為分散狀態。D-LFPΔN2漿料流動性提升也是如此。

圖1 64.1%(質量分數,下同)的D-LFP漿料和G-LFP漿料的流變特性[25](a)黏彈性圖,插圖為D-LFP(左)和G-LFP(右)漿料的形態;(b)黏度變化圖,插圖為D-LFPΔN2(左)和G-LFPΔN2(右)漿料的形態Fig.1 Rheological properties of slurries containing 64.1% (mass fraction, the same below) D-LFP and G-LFP[25](a)viscoelastic properties, inset shows pictures of aqueous slurries of D-LFP (left) and G-LFP (right);(b)viscosity properties, inset shows pictures of aqueous slurries of D-LFPΔN2 (left) and G-LFPΔN2 (right)
LFP顆粒表面的碳衍生物還會影響電池的電化學性能。顆粒表面的衍生物越多,包覆碳上的不能導電的sp3鍵合碳越多。G-LFP材料制備的電池的阻抗比D-LFP材料制備的電池的阻抗更高是因為G-LFP的電荷轉移阻抗更高。G-LFP更高的阻抗使得G-LFP有著更低的放電平臺和更低的比容量。
活性材料顆粒的尺寸大小對漿料的分散性和穩定也有著重要的影響。Bauer等[14]以顆粒尺寸分別為130 nm的LiFePO4(LFP)和8.9 μm的鎳鈷錳酸鋰(Li(Ni,Mn,Co)O2, NMC)為活性材料研究正極漿料的流變性和穩定性。
LFP漿料存在較為明顯的剪切變稀行為和屈服點,即LFP漿料形成了典型的凝膠類型結構。這是因為130 nm LFP顆粒具有大量的相互作用位點,特征直徑在100~200 nm的聚偏氟乙烯(polyvinylidene fluoride, PVDF)分子鏈形成了更平坦的構型,降低與其他顆粒間直接接觸的可能性。在漿料分散體中由PVDF和固體顆粒之間的吸引力形成了凝膠網絡結構。
和細小的LFP顆粒相比,具有相同固含量、由顆粒尺寸更大的NMC制備的漿料有著更低的黏度,僅存在輕微的剪切變稀行為,且無屈服點。這是流體系統的典型行為。將NMC體積分數升至30%時,該漿料的黏度會顯著升高,但仍沒有屈服點,表明該漿料也是典型的流體性系統。這是因為NMC顆粒尺寸為8.9 μm,PVDF分子特征尺寸遠小于NMC顆粒尺寸,很大一部分的黏結劑會沉降在與下一個顆粒距離很遠以至于不能形成橋接作用的位置,無法形成有吸引力的網絡結構。最終無法形成穩定的聚合物凝膠結構。當NMC顆粒增多雖能讓結合鍵數有所增加,NMC顆粒網絡結構的整體吸引力相互作用有所增強,但在低剪切速率下也能誘導結構分解,無法形成穩定的凝膠。由弱吸引力形成的凝膠網絡可以固定顆粒以實現顆粒系統的穩定均勻化,但必須適當調整吸引力,既要防止因形成過于堅固的凝膠網絡而無法達到電極涂覆過程中完全流化的要求;也要避免所形成的凝膠結構強度過弱而無法抵抗離散粒子的沉降。
2.2 黏結劑對漿料流變性的影響
黏結劑含量對漿料的流變特性有著重要影響。Li等[26]研究了黏結劑丁苯橡膠(styrene butadiene rubber, SBR)和羧甲基纖維素(carboxymethyl cellulose, CMC)的總含量恒定條件下不同比例的SBR/CMC對鈷酸鋰(LiCoO2)正極漿料流變性的影響。CMC在水溶液中解離從而形成陰離子電荷,CMC的特定吸附可以增加LiCoO2的負電荷密度和Zeta電位的大小,這為LiCoO2在水懸浮液中的分散提供靜電穩定機制。定義SBR在SBR/CMC中的占比為[SBR]。如圖2(a)所示,[SBR]為90%和70%時漿料的黏度曲線都比較復雜,在低剪切區域呈現剪切變稀,在高剪切率下呈現剪切增稠。隨著[SBR]的減小,漿料的表觀黏度增大,剪切變稀現象更明顯。圖2(b)中的相對黏度排除了SBR和CMC的流變本質特性對漿料流變的影響。很顯然,漿料的相對黏度隨著[SBR]的減小而降低,表明[SBR]的下降有利于漿料中電極顆粒的分散。此時仍可觀察到[SBR]較高時漿料存在復雜的流變行為,表明漿料中的電極顆粒同時存在著軟團聚體和硬聚集體。當[SBR]為50%時,漿料僅表現出剪切增稠行為。當[SBR]為30%和10%時,漿料表現出牛頓流體行為,漿料中的固體顆粒分散性能優良。
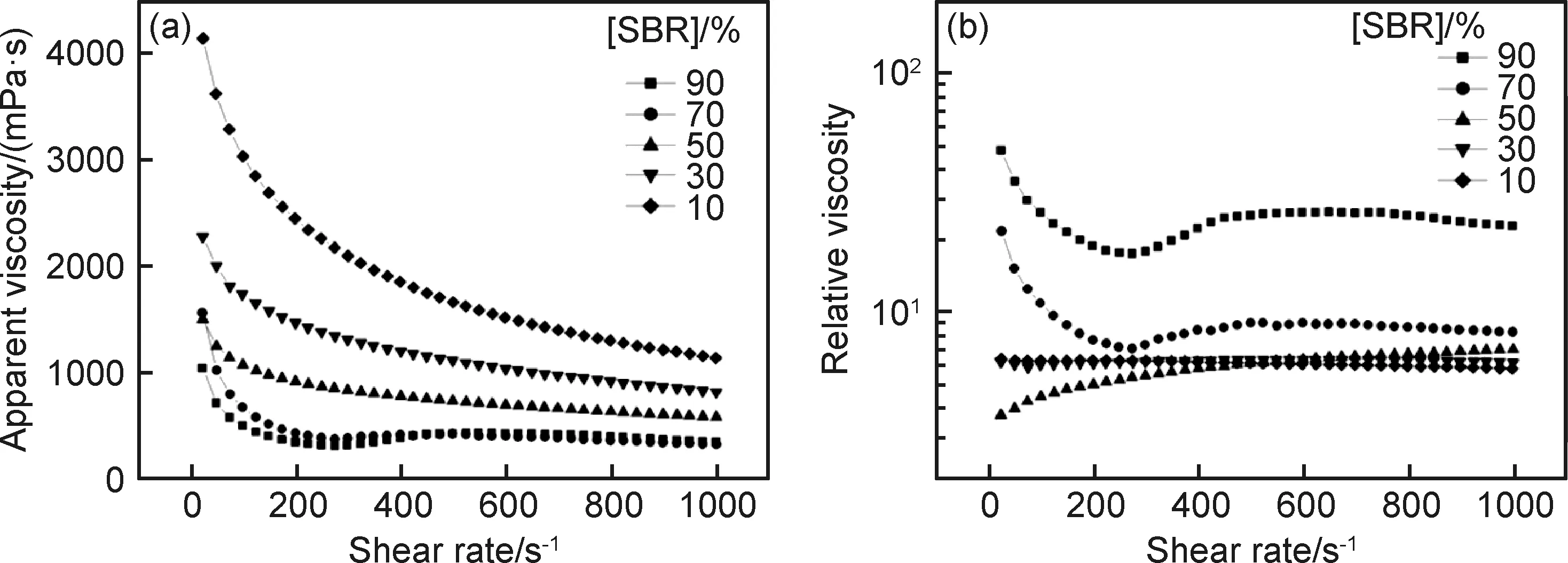
圖2 不同SBR含量下68%LiCoO2漿料的流變特性[26](a)表觀黏度;(b)相對黏度Fig.2 Rheological properties of slurries containing 68%LiCoO2 under different SBR contents[26](a)apparent viscosity;(b)relative viscosity
電極顆粒上的黏結劑吸附量隨著[SBR]減少而降低,有更多自由的黏結劑在干燥過程中隨著水分子遷移到電極表面。因此,當[SBR]減少時,電極中黏結劑分布的均勻性會下降,極片頂部(95%)和底部(5%)位置黏結劑含量的差異增大。黏結劑分布均勻性的降低導致極片的黏附力降低,即[SBR]較低時,極片較差的黏附性主要歸因于電極片中黏合劑分布均勻性的降低。
在電性能方面,決定電極表面電阻的主要因素是黏合劑的分布和電極黏合的性質,而不是電極粉末的分散。因此,當[SBR]降低時,盡管電極粉末的分散變得更好,但未壓縮電極片的表面電阻增加。在電化學性能方面,未輥壓的電極隨著黏合強度的降低總體上表現出容量下降的趨勢,但[SBR]為30%和10%的電極因其電極粉末的分散性極其優良,而不遵從這一規律。相反地,輥壓前容量最低的[SBR]為50%和70%的電極具有最高容量,這應當與它們在輥壓后顯示的最佳孔隙率相關。
此外,Lim等[27]報道了負極漿料的微觀結構對干燥過程中膠乳遷移的影響。負極漿料中的微觀結構取決于CMC和石墨的比例。當CMC與石墨的比例低時,由于石墨表面上吸附的CMC的空間排斥,石墨顆粒分散在漿料中。由于在干燥期間膠乳遷移,根據漿料的微觀結構所形成的膜的微觀結構不同。
黏結劑的濃度和種類均會影響漿料的流變性能和微觀結構。Lim等[33]研究了SBR,CMC黏結劑對石墨負極漿料三種體系流變性的影響。對于石墨-SBR體系,不含SBR的石墨顆粒漿料存在著屈服行為、在低剪切應力下黏度保持恒定,表明漿料顯示出類似固體的行為。經過某一臨界點之后黏度急劇下降,該臨界點即為屈服應力[34]。另外,G′>G″,且模量保持頻率獨立性。這一特性是由石墨顆粒的疏水性使其在水溶液中聚集,形成凝膠結構所導致[35]。加入少量SBR,黏度和模量均略微降低,表明隨著SBR加入漿料中凝膠強度降低,但其凝膠結構仍然存在。當SBR含量為15%時,漿料的屈服行為消失并存在剪切變稀行為;而當G″>G′時,模量值隨時間變化明顯。這均表明漿料中的凝膠結構消失。石墨顆粒漿料在高濃度SBR的分散作用下形成液體特性。
當SBR含量僅為3%時,石墨之間形成聚集的網絡連接,SBR顆粒被吸附在石墨顆粒表面上。當增加SBR含量到30%時,石墨顆粒表面和介質當中都存在著SBR。將SBR加入漿料中,SBR吸附在石墨表面上并降低石墨顆粒之間的吸引力。SBR本身不帶電荷,但存在著攜帶陰離子電荷的表面活性劑;表面吸附了SBR的石墨通過靜電排斥作用降低顆粒間的疏水吸引力而得到分散。
對石墨-CMC體系的流變性能研究發現,當CMC含量由0%增加至0.1%時,石墨漿料的黏度、屈服應力、模量都會有一定程度的降低,但其屈服特性仍存在,正如之前在石墨/SBR部分提到的一樣。CMC是可以在水溶液中電離成鈉陽離子和聚合物陰離子的聚電解質,這些離子通過靜電力相互作用影響聚合物構象。CMC的特定吸附增加了石墨顆粒表面的負電荷密度和Zeta電位的大小,這為石墨顆粒在水懸浮液中的分散提供靜電穩定機制。CMC由0.4%增至1.0%時的漿料的屈服現象消失并出現剪切變稀行為。此時CMC在石墨表面上的吸附量進一步增加,石墨顆粒通過吸附的CMC之間的靜電排斥而得到良好的分散。由CMC水溶液的比黏度隨其濃度的變化情況可知,CMC存在3個轉變臨閾值,可知此濃度范圍內的CMC聚合物鏈形成纏結。此時漿料的黏度和模量隨著CMC濃度的增加而增大是由形成纏結的CMC所導致的。當CMC由1.4%增加到1.7%時,漿料中屈服點再次出現,這表明此時石墨漿料中重新出現凝膠結構。
由漿料和CMC溶液的tanδ隨CMC濃度的變化情況(其中tanδ=G″/G′,頻率為0.1 rad/s)可知,在CMC含量低于0.28%時,tanδ<1,表明漿料為固態特性;超過該臨界濃度0.28%后,tanδ>1,表明漿料呈液態特性。CMC分子被石墨顆粒表面所吸附,且吸附量隨著CMC的增加而增加[27,36],此時漿料的黏度和損耗/剪切模量都會下降。CMC含量超過下臨界濃度(約為1.3%)之后,tanδ再次小于1,這與CMC體系漿料在該濃度之后再次出現屈服現象相吻合。而在下臨界濃度之后漿料中出現的凝膠結構是由CMC分子形成的聚合物網絡結構所導致[37]。
在石墨-CMC-SBR漿料中,當僅有0.7%CMC時的漿料存在屈服現象和頻率獨立的模量。加入SBR后,漿料的黏度和模量均有所下降。此時CMC對石墨的吸附量非常小,SBR可以吸附在石墨表面,吸附在石墨表面上的SBR能夠起到分散石墨顆粒的作用。CMC含量增至0.7%的漿料的黏度和模量幾乎不隨著SBR含量的增加而發生改變,SBR僅存在于石墨顆粒之間,而不是在石墨顆粒表面。這表明當漿料中同時存在CMC和SBR時,CMC比SBR更優先地吸附在石墨表面上,并且它在分散石墨顆粒中起主導作用。這與Li等[38]研究混合步驟對黏結劑性能影響的結果一致。當漿料中的CMC濃度足夠高時,CMC對漿料微觀結構的形成起主導作用。
2.3 導電劑對漿料流變性的影響
鋰離子電池中存在離子傳導和電子傳導兩種導電方式。導電劑越多,電子導電性越強。但體積有限的鋰電池中活性材料將會減少,使得電池的容量降低。Cheon等[18]的研究表明電池容量與倍率性能和導電劑含量的對應關系是呈現相反趨勢。
電極中電子傳導可以用滲透理論來解釋[39]。在該理論中,導電物質作為連續相,但相間存在電阻,而導電劑的形貌千變萬化。因此,導電劑的形貌對于漿料性能的影響至關重要。Takeno等[40]研究了常用的乙炔黑(acetylene black, AB)、石墨以及氣相生長碳纖維(vapor grown carbon fiber, VGCF)的形貌對于漿料流變性的影響。AB由納米尺寸的碳顆粒組成,彼此連接,通過聚集和團聚作用形成簇狀物;石墨呈鱗片狀,VGCF為纖維狀,且發生纏結。
隨碳含量的增加,AB和VGCF漿料的電阻率隨著碳含量的增加而降低,而石墨漿料的電阻率基本不變;含相同碳含量的AB和VGCF漿料的電阻率基本一致,且小于石墨漿料。漿料的穩流黏度與結構黏度有關;G′和G″與漿料中的分散結構有關。分析這三種導電劑漿料的流變特性發現,AB和VGCF漿料的黏度均隨著含碳量的增加而升高,黏度值在低剪切速率更高,表明這兩種漿料內部的網絡結構隨著含碳量的增加而增加,并且都是非牛頓流體。其中AB漿料中的G′大于G″這一特性表明漿料為類固體性質。此外,具有2%和3% AB的電極漿料在低角頻率區域中的G′顯示出的平穩區域,這一現象表明電極漿料中AB網絡結構的生長。VGCF漿料的G″大于G′的結果表明VGCF漿料為類液體性質。石墨漿料的黏度值幾乎不隨剪切率變化,且黏度值隨含碳量變化不明顯,表明石墨漿料是牛頓流體;漿料內部幾乎沒有網絡結構。造成這些差異的原因在于三種導電劑的形貌不同。點狀的AB碳顆粒在漿料中會發生嚴重的聚集和團聚,形成彼此連接的剛性的電子傳導網絡;并且顆粒間的聚集和團聚作用隨著碳含量增加而顯著升高,從而AB漿料形成凝膠結構,其電子傳輸路徑優良。纖維狀的VGCF存在著能夠包含溶劑存在的空隙,這使得VGCF漿料表現為液態特性;線狀的VGCF能夠發生纏結,并且纏結程度隨著碳含量的增加而顯著升高。從而VGCF漿料為非牛頓流體,其中有著相對較好的電子傳輸路徑。片層狀的石墨的顆粒尺寸相當大,并且是通過顆粒間點接觸來形成電子傳導路徑。故其電子傳導路徑的量不隨碳含量的增加而增加。經分析三種漿料制備的電極的電阻值可知,具有電子傳導網絡結構的電極漿料呈現的類固體特性有利于獲得具有低電阻的極片。
2.4 溶劑對漿料流變性的影響
種類繁多的黏結劑的親水性能存在差異。根據黏結劑的親水性來選擇適合的溶劑可以提高漿料的分散均勻性。溶劑的種類會對漿料的流變性產生影響。Li等[41]比較了水基漿料和有機基漿料的分散均勻性。水基漿料的溶劑和黏結劑分別為去離子水和SBR,并以CMC作為SBR的增稠劑;有機漿料中的溶劑和黏結劑分別為甲基吡咯烷酮(NMP)和聚偏氟乙烯(PVDF)。兩種漿料的活性物質和導電劑均分別為LiCoO2和石墨。
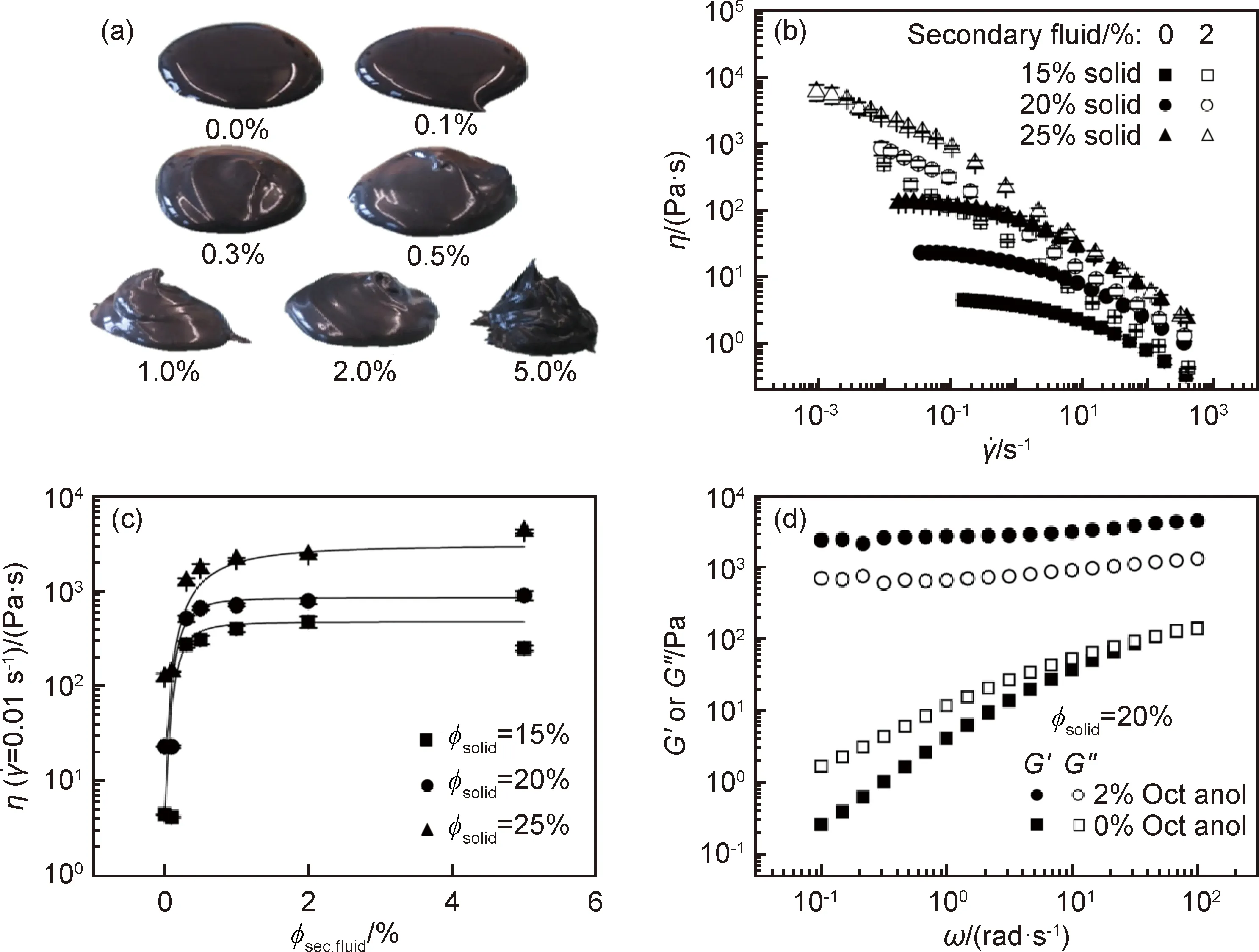
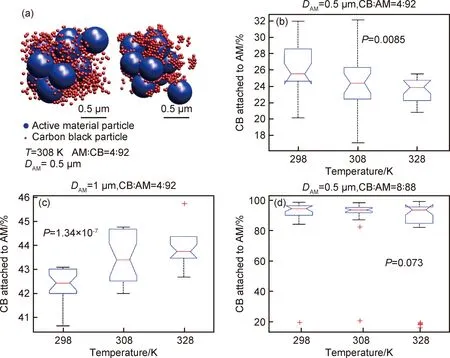
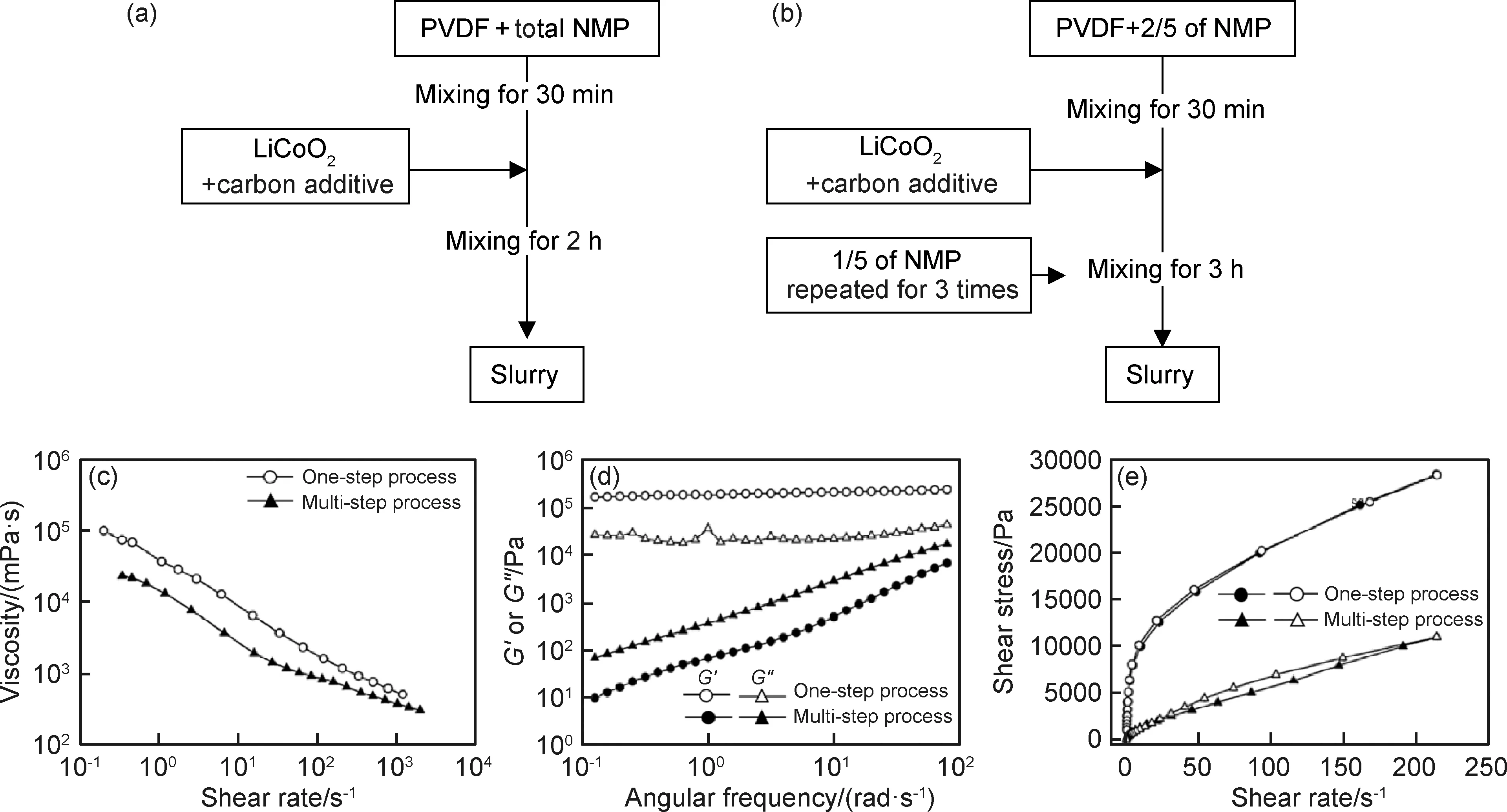
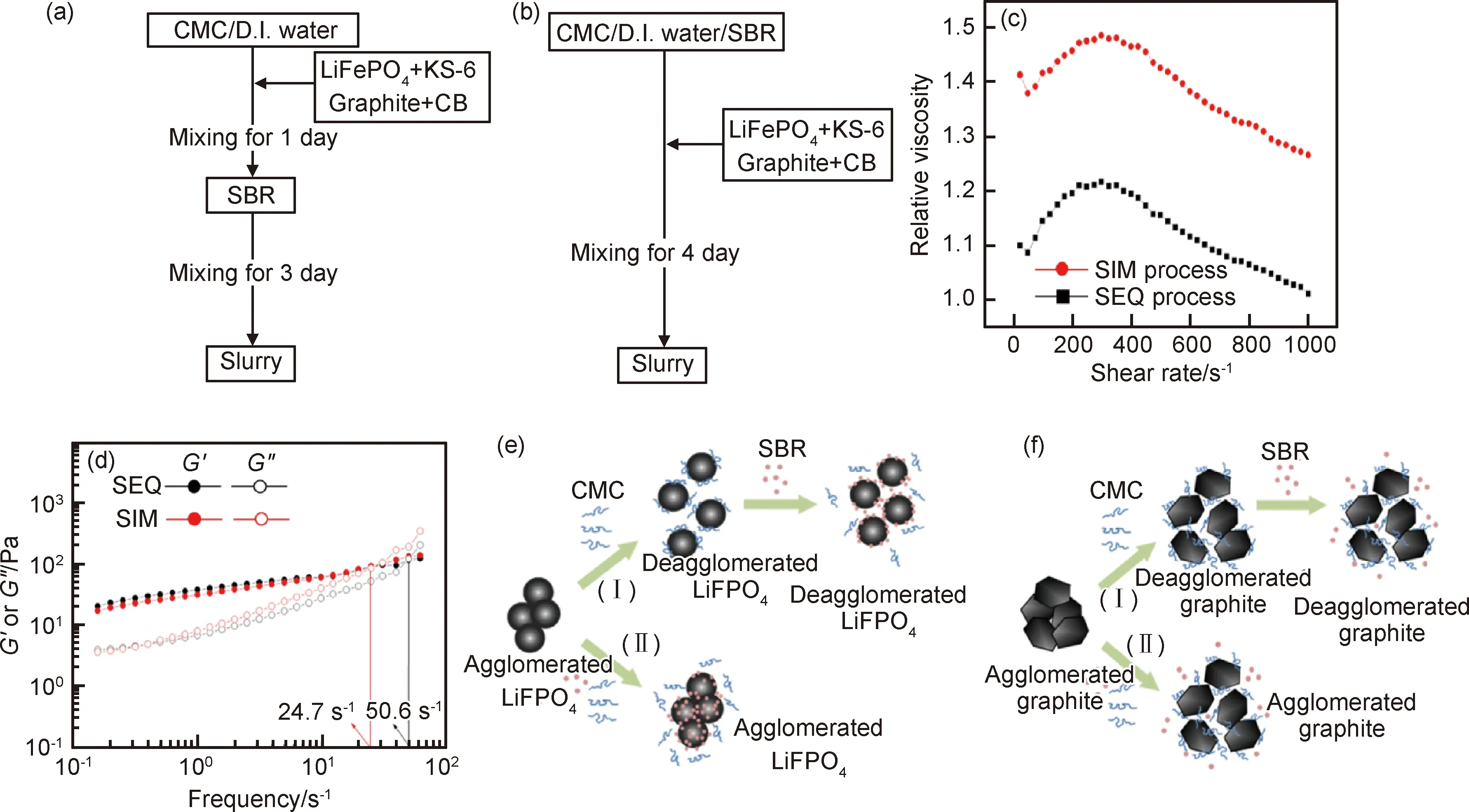
由Herschel-Bulkley方程[42]σ=σy+ηγn對漿料的流變性能進行分析可知,對于水基漿料,n=0.52且σy=25.5 Pa,而有機基漿料的n=0.84且σy=9.5 Pa。相對于水基漿料,有機基漿料具有更大的n、更小的σy和較小的磁滯回線面積,可知有機基漿料的分散性更好,該結果從有機基漿料具有更低的G′和G″也得以體現[23-24]。而水基漿料中的G′對頻率更低依賴性則表明該漿料呈現出的結構更加偏向于固體狀。水基漿料的G′>G″而有機基漿料的G′ 有機基漿料更好的流動性會使得該漿料中的石墨更容易與溶劑一起流到干燥電極片的頂部,從而造成電極片成分不均勻,另一方面,由水基溶劑漿料制備的電極片中成分的分布會相對更均勻,這很少在文獻中有報道[41]。在有機基電極片的頂層上可觀察到大量的石墨,其幾乎覆蓋了其他成分,而在其底層發現石墨含量很少。對于水基電極片,頂層和底層的石墨含量之間的差異不太明顯。主要是因為有機漿料的低黏度和高流動性使得石墨在干燥過程中重新排列。并且有機基電極片的成分不均勻分布會使其電阻和充電放電效率方面的性能比水基電極的性能差。 在電極的電學和電化學性能方面,未輥壓的有機基電極因其黏結劑分布不均勻、電阻高,導致對應電池的電壓平臺相對更低。輥壓后,有機基電極的電池顯示出比具有水基電極的電池更大的平臺電壓位移,這表明輥壓階段對于有機基電極的電化學性質尤其重要。此外,溶劑含量對漿料的流動特性和分散也有著十分重要的影響。Ligneel等[44]研究發現:當溶劑濃度低于最佳濃度時,漿料表現出屈服應力,可抑制流動并防止內部各組分均勻分布;溶劑濃度高于最佳濃度時,釩酸鋰(Li1,1V3O8)和炭黑顆粒在低黏度漿料中發生沉降。 溶劑濃度較高的漿料分散體有著很低的屈服應力并表現出牛頓行為,溶劑濃度相對較低的漿料有著更高的屈服應力并表現出剪切變稀行為。這是典型的絮凝分散體漿料,其中存在于顆粒之間的弱吸引力形成了絮凝物的顆粒簇。超過臨界濃度時,絮凝物在整個漿料體系中相互連接,從而形成薄弱的網絡[44]。屈服應力便是漿料中存在網絡結構的體現。隨著溶劑濃度的降低,整個漿料系統的連通性和其屈服應力都會增加。相反,在稀釋的漿料分散體中,絮凝物以獨立個體存在,未能形成網絡結構,漿料保持液體特性,且無屈服應力。 已有報告研究了絮凝分散體漿料的沉降與其濃度和容器尺寸之間的關系[45],發現絮凝物具有一定的機械強度,故能在由弱剪切力和重力沉降所導致的碰撞中保持其特性。而在低剪切速率下,絮凝物十分容易聚集形成絮狀物簇,這些絮凝物可延伸到容器壁并使得分散體漿料具有塑性和類固體性質。對于稀釋的分散體漿料,其中的絮凝物或絮凝物簇作為單獨的單元而不是鏈或網絡發生沉降,并且漿液表面和上清液之間的距離Y(t)隨時間線性降低。當溶劑含量較低時,其中的絮凝物沉降為連貫的網絡,且初始沉降速率(Y(t)曲線的斜率)非常低,然后隨著時間的推移而增加[45]。Ligneel等[44]的研究發現,對于溶劑濃度高于0.004 mL/mg的所有漿料,其行為明顯“稀釋”分散,并與其流變學測量結果中沒有明顯屈服應力的結果一致。相反,在溶劑濃度為0.004 mL/mg的漿料中幾乎沒有檢測到沉降,因為漿料中的顆粒已經形成具有較強的屈服應力的連貫網絡結構。 顆粒之間的弱吸引力有利于抵抗由最重的活性材料和導電劑附聚物產生的集體沉降。對于所有固液分散體而言,顆粒間吸引相互作用的發生并不普遍,但這卻是低濃度和非極性溶劑中的懸浮漿料的常見特征。相反,在極性溶劑中,漿料的分散顆粒通常通過靜電排斥力達到穩定,但卻很可能使得較重顆粒和較輕顆粒之間發生分離。 分散添加劑在漿料的制備過程中對顆粒團簇的分布狀態有著重要的影響。Bitsch等[46]提出了一種通過毛細管力來控制漿料流變性能的概念。在漿料中采用CMC水溶液作為溶劑,加入少量的與水不相溶的正辛醇,利用CMC水溶液與正辛醇對漿料內部固體顆粒間接觸角的不同,使得漿料內部產生毛細管力,進而控制漿料的流變性能和懸浮穩定性。 石墨顆粒、水和正辛醇三者之間的三相接觸角小于90°,正辛醇會優先潤濕石墨顆粒,形成貫穿整個漿料體系(sample-spanning)的網絡結構。如圖3(a)所示,沒有添加正辛醇和添加極少量的正辛醇的懸浮液像稀懸浮液一樣擴散。正辛醇含量的繼續增大導致漿料的紋理發生劇烈變化并產生似糊狀的行為,這是因為正辛醇所產生的毛細管力引起的粒子網絡(sample-spanning),導致低剪切下的黏度急劇增加。隨著正辛醇添加量進一步增大,懸浮液變得越來越凝膠狀或糊狀,這意味著其沉降穩定性得到改善。 圖3 不同含量正辛醇下漿料的流變特性圖[46](a)負極漿料在不同含量正辛醇下的特性(固含量φtotal為20%(體積分數,下同));(b)黏度vs剪切速率曲線;(c)低剪切黏度曲線(帶誤差棒的符號);(d)含有2%正辛醇的漿料和不含正辛醇的漿料在恒定固體部分(固含量為20%)下的儲存和損耗模量Fig.3 Rheological properties of slurries under different 1-octanol contents[46](a)characteristic of anode slurries with solid content of 20% (volume fraction, the same below) under different 1-octanol contents; (b)viscosity curves vs shear rate;(c)low shear viscosity curves (symbols with error bars); (d) storage and loss modulus for the slurry containing 2% 1-octanol and the slurry without 1-octanol at a constant solid content (20%) 漿料黏度隨著固含量的增加而上升,且都存在著剪切變稀行為(圖3(b))。向懸浮液中加入2%的正辛醇(secondary fluid, sec.fluid)會形成由毛細力引起的強網絡結構,從而導致低剪切速率下的黏度會急劇增加。而在高剪切速率下,毛細管網絡被破壞,故此時這兩類漿料的黏度是相似的。低剪切速率下的黏度隨著正辛醇含量的增加而急速上升。含0.5%正辛醇的漿料達到低剪切黏度的平臺值,表明此時網絡形成完成。進一步添加正辛醇不會改變結構和流動性質。在無正辛醇的漿料中,模量具有頻率依賴性,并且G″>G′。該漿料為良好分散的溶膠[21]。添加2%正辛醇后,漿料的模量呈現出頻率無關性,且G″占據主導,即漿料表現出凝膠狀行為。加了正辛醇的電極邊緣形狀平緩很多,并且厚度增加得更快,主要是因為低剪切區域中的高黏度值阻止了由重力或表面張力引起的擴散和流動,因此獲得良好的輪廓精度。 Li等[28]以聚醚酰亞胺(polyetherimide, PEI)為分散添加劑,研究了分散劑含量對LiFePO4正極活性材料漿料的影響。發現所有的漿料都存在剪切變稀行為。加入黏結劑后漿料內部顆粒之間的連接變強,漿料的屈服應力大大增加。含1.5%和2.0% PEI的漿料表現出最低的屈服應力和最大的冪律指數,產生最高質量的漿料。Jung等[13]為了減輕含水黏合劑體系中的碳聚集問題,將乙醇用作漿料的分散添加劑。發現乙醇的添加使得碳顆粒能夠更均勻地混合在漿料中。 總體來說,分散添加劑對漿料的分散均勻性和沉降穩定性的影響方式主要是改變溶劑和顆粒表面的接觸角和通過靜電作用力阻礙團簇的合并。顆粒表面和溶劑之間的接觸角越小,溶劑對顆粒表面的作用力就越大,從而使得粉體的分散效果更好[47]。分散添加劑能夠降低這兩者的接觸角、減小團聚體內部作用力和顆粒-溶劑作用力之間的差值,有利于團聚體的分散[11,48-51]。但分散添加劑并不能使三相接觸角為零,故粉體的分散仍需要輸入其他方式的能量才能達到最佳效果。在漿料中,團簇的尺寸和形貌、活性物質和導電劑的分布,都受到相互碰撞的團簇的重組過程控制[28,52-55]。陰離子或陽離子分散添加劑通過靜電作用力來改變團簇內部的作用力,阻礙團簇合并[51,56-58]。分散添加劑的選擇應避免其在干燥電極中的殘留和對鋁集流體的腐蝕[59]。 Han等[60]通過在聚丙烯酸(polynuclear aromatic hydrocarbons, PAH)中添加不同量的氫氧化鈉(NaOH)制成不同pH值的黏結劑來制備負極電極漿料,PAH的pH值隨NaOH含量的增加而非單調遞增式地增加。溶液中聚丙烯酸酯鏈的結構與黏度的變化密切相關。PAH溶液顯示弱酸性,因此溶液中沿聚合物鏈的未離解的羧基彼此締合以形成氫鍵。每個聚丙烯酸酯分子形成隨機卷曲的結構。PAH溶液顯示出低黏度。添加NaOH之后,PAH的電離度增大,導致相鄰離解的羧基之間由于靜電作用相互排斥,聚丙烯酸酯鏈被拉伸,導致溶液黏度增加。黏結劑黏度的增加有利于電極內部活性物質的均勻分布。含PAH黏結劑的電極中存在著硅和石墨的聚集,而在被中和后的PAH黏結劑中,這些顆粒得到很好的分散性。 此外,Hu等[61]也研究了聚丙烯酸(polyacrylic acid, PAA)在石墨-硅復合電極的循環性能和漿料穩定性之間的平衡。用LiOH調節PAA水溶液的pH值,發現PAA的pH值隨LiOH增加的變化情形與PAH的pH值隨NaOH含量的變化情況類似,其中PAA黏合劑羧基中的H+/Li+交換作用稱為PAA的預鋰化。由預鋰化PAA溶液的表觀黏度與剪切速率的關系發現在低剪切速率下,隨著PAA鋰化程度的增加,PAA溶液的黏度急劇增加,這可歸因于顆粒的遷移率降低。可以預期在較高的pH值下PAA中羧基的離子化程度更高,因此它們的遷移率受到很大限制,可能導致黏度大大提高[62]。此外,所有溶液中都存在著剪切稀化。在低剪切速率下較高的連續相黏度可減緩懸浮顆粒的沉降和聚結來提高水性懸浮液的穩定性,而在高剪切速率下發生的剪切稀化使漿料在制備過程中更容易混合,這對于生產高質量層壓板很重要。因此,在低剪切速率下足夠高的黏度與剪切稀化相結合是Si納米顆粒在高濃度懸浮液中共混以進行大規模層壓工藝的理想組合。通過對比PAA-6.0和PAA-2.1黏合劑的漿料的流變特性發現,與PAA-2.1漿料相比,PAA-6.0漿料在低剪切速率下具有十倍的高黏度(層壓過程中具有更好的漿料穩定性),但是在剪切速率超過20 s-1時,兩種黏度變得相近。因此,從批量加工的角度來看,鋰化的PAA-6.0黏合劑的確比未處理的PAA-2.1黏合劑具有優勢。 但是,從材料化學的角度來看,PAA黏合劑的預鋰化可能是有害的,因為它不僅會導致電極基質中顆粒之間的黏合/內聚力降低,還會腐蝕硅顆粒。因此,PAA黏合劑的預鋰化會導致半電池和全電池的循環性能變差。當pH值超過7時,這種不利現象將變得更加明顯。 在鋰離子電池漿料中,活性材料(active material, AM)一般都是由很多小顆粒組成的大顆粒(二次顆粒),這些二次顆粒的尺寸大約在幾十到100 μm,而小顆粒尺寸約在1 μm左右。導電劑顆粒尺寸更小,在納米級別。在這樣的體系中,電極的質量會受到諸多因素的影響,漿料內物質之間可能會受到黏性力、布朗運動等的影響而影響其分布,這其中溫度對于細小顆粒布朗運動的影響尤為顯著。Zhu等[63]通過布朗動力學仿真研究了溫度對正極漿料內導電劑在活性物質上的吸附量的影響,如圖4(a)所示。在該模擬中,炭黑(carbon black, CB)通過吸引力被吸附在AM表面;在CB團簇中,CB之間存在著短程的吸引力。整個體系的穩定性同時受到范德華力、靜電力的影響。對于這些微小顆粒組成的體系,布朗運動對于整個體系的狀態影響很大。 Zhu等[63]發現當AM粒徑為1 μm時,隨著溫度的升高,AM表面吸附的CB含量增加,但當AM粒徑為0.5 μm時則呈相反的趨勢。圖4中P值取決于CB的吸附對溫度的依賴性,依賴性越弱,P值越大。Zhu等[63]認為在粒徑較大的體系中,溫度越高,CB顆粒的布朗運動越劇烈,越容易克服勢壘達到穩定狀態使得總體的能量保持最低,此時AM粒子和CB粒子之間的吸引力將足夠大以降低由布朗運動引起的脫離的可能性。而對于粒徑較小的體系,CB之間及CB與AM之間的吸引力較弱,在整個體系內,相比于其他的作用力,布朗運動更為突出,布朗運動劇烈之后容易破壞原本體系內的網絡結構,使得CB的吸附量降低。 圖4 吸附在AM顆粒上CB含量隨溫度的變化情況[63](a)布朗動力學仿真漿料內結塊情況;(b)~(d)吸附在AM顆粒上的CB的含量隨溫度的變化Fig.4 Percentage of CB attached on AM vs temperature[63](a)aggregated structure after Brownian dynamic simulation;(b)-(d)percentage of CB connected to AM cluster plotted against temperature 漿料中的液體組分擴散到固體顆粒的內部區域孔隙中,不同的混合條件可以引起液體吸收程度的偏差。粉末的不同比表面積和液體吸收程度會加大這種差別[20]。據報道,在漿料的制備過程中,即使使用相同種類和數量的導電劑、活性材料、黏結劑和溶劑等原材料,不同的混合順序會對漿料的性質及電極的性能都產生影響[16,20-21,38,64-69]。 溶劑添加的方式會對漿料的性能產生重要影響。Lee等[21]研究了溶劑混合順序對LiCoO2/導電劑/PVDF漿料性能的影響,以N-甲基吡咯烷酮(N-methylpyrrolidone, NMP)為溶劑,比較了兩種不同的制備步驟順序,具體情形如圖5所示。 圖5 兩種混合步驟下漿料的制備示意圖和流變特性圖[21](a)一步法;(b)多步法;(c)黏度隨剪切速率的變化;(d)黏彈性模量隨角頻率的變化;(e)兩種制備步驟下漿料的流動性能Fig.5 Schematic diagrams for two kinds of slurry preparation methods and the corresponding rheological properties[21](a)one-step process;(b)multi-step process;(c)viscosity vs shear rate;(d)viscoelastic modulus vs angular frequency;(e)flow curves of the slurries prepared by two mixing processes 由圖5(c)中漿料的黏度曲線可知,兩種漿料都存在剪切變稀的情況;且穩定性都比較好,固體成分的沉降不明顯。在圖5(d)中可觀察到,一步法所制得漿料的模量與頻率無關,且G′>G″,表明該漿料為凝膠態,漿料內部的固體顆粒彼此連接形成內部網絡[70],這從圖5(e)中也可得到確認。這主要是因為在混合的初始階段,一步法漿料處于黏度相對較低的狀態,所施加的較低剪切速率未能把漿料中的網絡結構破壞。多步法漿料中模量值與頻率相關、G′ 多步法漿料中網絡結構的破壞在圖5(e)中所示的滯后流動曲線中得到進一步的證明。通過增加剪切速率得到剪切應力值與通過降低剪切速率獲得剪切應力值的差異是由剪切引起的網絡結構破壞導致的[71-72]。 此外,多步法制作的電極AM和碳顆粒分布更均勻,使得該電極的極化更微弱。與一步法制備的電池相比,多步法制備的電池的循環性能和倍率性能更好,這與Kim等[20]研究的結果相吻合。 黏結劑的添加順序也對漿料的分散性和穩定性有著重要的影響。Li等[38]研究了SBR和CMC對水系磷酸鐵鋰漿料性能的影響,溶劑為去離子水(D.I. Water)。比較了兩種不同的制備順序:第一種為先添加CMC后加入SBR的順序步驟,即按順序添加(sequence, SEQ)過程;第二種為同時加入SBR和CMC的步驟,即同時添加過程(simultaneously, SIM)過程,如圖6所示。 與SBR混合可降低LiFePO4和KS-6石墨的沉降高度,表明SBR在分散這兩種粉末方面具有一定的效率。當與CMC混合時,LiFePO4和KS-6石墨的粉末懸浮液的分散穩定性優異,它們都難以完全沉降并且在上清液和沉降的粉末之間分界面不清晰。這些結果表明CMC分散這兩種粉末非常有效。 相對黏度定義為電極漿料的表觀黏度與CMC水溶液的表觀黏度之比。由圖6可知,兩種漿料都存在著復雜的流變性能:低剪切率下剪切增稠,高剪切率下剪切變稀。而由SEQ漿料的相對黏度更低可知該漿料的團聚程度相對較低。兩種漿料G′>G″且模量均與頻率相關,這兩種漿料都更偏向于類固體結構。但SIM漿料的弛豫時間相對更低,該漿料的分散性相對更好。 對于LFP,不論是何種混合步驟,SBR都會比CMC優先吸附在LFP上面。相反地,石墨和碳則會優先與CMC發生吸附。CMC為可分離的聚合物電介質,其特性吸附能為活性顆粒同時提供空間位阻和靜電排斥力,即CMC的分散機制屬于電位穩定效應。因此,SBR和CMC的添加順序會對活性顆粒的分散效果產生重要影響。SEQ過程中吸附的CMC引起的電位穩定效應可以使LFP有效的解聚集。LFP上CMC的吸附被后面添加的SBR取代之后,LiFePO4也已經得到很好的分散。在SIM過程中,只有SBR會被吸附到LFP上。由SBR導致的空間效應并不能使LFP顆粒得到良好的分散,故此漿料中還存在著很大的LFP團聚體,具體機制如圖6所示。 圖6 水系LiFePO4電極漿料的制備過程、流變特性圖和分散機理[38](a)SEM過程;(b)SIM過程;(c)漿料的相對黏度隨剪切速率的變化;(d)G′與G″隨掃描頻率的變化;(e)LiFePO4在水懸浮液中的分散機理;(f)KS-6石墨在水懸浮液中的分散機理Fig.6 Fabrication process,rheological properties and dispersion mechanisms of the aqueous LiFePO4 slurries[38](a)SEQ processes;(b)SIM processes;(c)relative viscosity vs shear rate;(d) G′ and G″ vs sweeping frequency; (e)dispersion mechanisms of LiFePO4 in an aqueous suspension;(f)dispersion mechanisms of KS-6 graphite in an aqueous suspension 此外,相對于SIM電極,SEQ電極的氧化峰和還原峰更勻稱更尖銳,有著更高的密度,并且該電極的兩峰之間峰位差異更少,其循環穩定性更好。這都是因為SEQ電極中活性顆粒的解團作用更好,更小的顆粒尺寸縮短了鋰離子的擴散路徑;活性顆粒更好的分散作用為鋰離子的潛入和脫出能夠提供動力和可逆的電化學反應。顆粒的團聚使得SIM電極片有較高的電阻,也就是活性顆粒的分散和顆粒之間的接觸性越差,IR降越大,從而SIM電極片容量和平臺電壓更低。 由于范德華力或靜電吸引力以及重力作用的存在,漿料中的顆粒很可能會發生團聚或沉降。這大大影響了漿料組分的分散均勻性和漿料整體的抗沉降穩定性。在水系漿料中更容易存在相對較強的氫鍵和靜電力,這為顆粒的團聚提供了有力的條件。因此,水系漿料通常需要添加分散劑,以提供靜電屏蔽的方式來防止顆粒的團聚,使導電劑和活性物質分散得更均勻。此外,還可通過添加表面活性劑,以改變團簇內部的作用力來改變接觸角方式,提高水系漿料的均勻性。但表面活性劑可能在漿料干燥之后仍然存在于電極顆粒的表面,這不利于電極的電導性。故應選擇在干燥過程中容易揮發的表面活性劑。在油系漿料中,可以通過黏結劑的分子量和顆粒-黏結劑相互作用的強度,使得黏結劑的垂懸端相互纏繞成網絡,形成橋連絮凝。這些跨越體積的網絡有利于提高漿料的抗沉降性。而在水系漿料中則要避免形成這種聚合物網絡結構。 所制備漿料的性能還與其投料順序有重要關系。對于粉體顆粒的投放,多步添加制備的漿料中組分的分散比一次性添加制備的漿料中組分的分散更均勻。在水系漿料中最常用的黏結劑為CMC和SBR,這兩種黏結劑的分散能力存在著較大的差別。因此,黏結劑的混合順序對于水系漿料的影響十分明顯。CMC的分散能力高于SBR的分散能力。但對于磷酸鐵鋰,SBR的吸附競爭力遠高于CMC。因此,制備磷酸鐵鋰漿料時應該先放CMC后放SBR。而對于石墨類的負極材料,CMC的吸附競爭力高于SBR。因此,制備石墨漿料時這兩種黏結劑的投放順序影響不會很大,但SBR只在CMC含量很低時才能起到分散作用。 最后,其他一些因素也會影響鋰離子電池漿料的性能,如pH值、溫度。此時,應該考慮漿料中的AM的種類、導電劑的大小、黏結劑的分子結構來綜合進行選擇,以便制備出性能優良的漿料。 電極漿料的制備是鋰離子電池生產過程中的第一步也是最重要的一步,其分散均勻性和穩定性會對后續的涂布、干燥等一系列過程有著顯著的影響,進而決定了鋰離子電池的綜合性能。電極漿料是一個多組分經一定步驟混合而成的非平衡態體系。其存在的最大的問題便是內部顆粒由于靜電吸引力、范德華力或氫鍵等作用團聚,或是在重力作用下發生沉降,從而影響了漿料的分散均勻性和穩定性。因為電極漿料的物理特性,漿料的性能不易直接觀測,研究者們通常都是研究與其相對應的流變性能來間接研究漿料均勻性和穩定性。 研究表明漿料的性能嚴重依賴于原料組分之間的相互作用。總體而言,通常存在以下三種方法來提高漿料的性能。首先,漿料中的顆粒間存在著的靜電排斥作用力或空間勢壘,能有效地防止離散顆粒形成更大的團塊。如SBR,CMC會對負極顆粒產生特定吸附,吸附了SBR/CMC的負極顆粒通過靜電排斥作用力而得到很好的分散。由于黏結劑之間的吸附競爭力和分散能力存在著差異,因此要考慮黏結劑的投放順序,以便獲得性能更好的電極漿料。其次,改變漿料中顆粒的遷移率可以通過增加漿料的黏度來實現,如通過添加增稠劑能提高漿料的黏度。較高的黏度能夠降低顆粒的動能并阻止顆粒克服能量障礙,從而形成空間位阻降低顆粒的沉降速度。添加適量分散劑可以改變溶劑和顆粒表面之間的接觸角從而提升漿料的均勻性和穩定性,但該方法并不能十分有效地抑制大顆粒的沉降,并且一般要求所添加的分散劑不會對后續電極的電化學性能產生影響。降低漿料內部顆粒沉降速度更為有效的方法是要在漿料內部形成弱凝結網絡結構。此時,漿料中存在一個內聚的、顆粒間非直接連接的網絡,該網絡結構的體積足以延伸至整個容器。適宜的漿料能夠提供具有足夠吸引力的穩定性網絡以抵抗粒子的重力效應,并且該網絡有足夠的負載能力防止由于其自身質量而發生合并。該性能可使得漿料在低剪切速率下仍能流動,并避免流動的漿料內部網絡結構的重新形成。 然而,本文在對新型材料制備漿料的討論還存在不足,正極方面如高鎳正極材料、富鋰錳基正極材料,負極方面如新型的硅碳材料等。由于這些材料的漿料特性文章目前比較少,無法系統的總結。此外,導電劑的分散工藝仍需進一步的研究,特別是對于新型導電劑石墨烯、碳納米管等。 當前對于電極漿料的流變性能研較少,研究的重點更多的是在電極的電化學性能上。而電極優良電化學性能得以發揮的前提條件在于漿料的均勻性和穩定性。漿料的分散均勻性和穩定性受到其制備的整個過程的影響,包括原料組分和分散條件。這些外部的、內部的因素在漿料的制備中是相互關聯、相互制約、彼此影響的,類似于一個整體的有機系統。因此,在考慮所期望的電極電化學性能之前,應綜合考慮多孔電極的微觀結構和影響漿料性能的因素以及相互之間的關系。2.5 分散添加劑對漿料流變性的影響
2.6 pH值對漿料流變性的影響
2.7 溫度對漿料流變性的影響
2.8 混合步驟對漿料流變性的影響
3 提高電極漿料性能的策略
4 總結與展望
