油酸甲酯烯烴交叉復分解制備烯烴化學品中底物影響機制探究
2021-07-03 03:24:22舒恒毅鄭志鋒劉守慶何宏舟黃元波
林產化學與工業 2021年3期
關鍵詞:催化劑
舒恒毅, 鄭志鋒, 劉守慶, 何宏舟, 黃元波,3*
(1.西南林業大學 材料科學與工程學院,云南 昆明 650224; 2.集美大學 機械與能源工程學院,福建廈門 361021; 3.林業生物質資源高效利用技術國家地方聯合工程研究中心;西南地區林業生物質資源高效利用國家林業和草原局重點實驗室,西南林業大學,云南 昆明 650224; 4.廈門市現代農業生物質高值化技術重點實驗室(廈門大學);福建省生物質高值化技術工程研究中心(廈門大學);廈門大學 能源學院,福建 廈門 361102)

1 實 驗
1.1 材料、試劑與儀器
油酸甲酯(MO)(純度99%)、烯丙基三甲基硅烷(純度98%)、二氯甲烷(純度99.9%)、2-甲氧基丙烯(純度95%)、烯丙基縮水甘油醚(純度99%)、苯乙烯(純度99%)、乙酸烯丙酯(純度99%)、丙烯腈(純度99%)、丙烯酸甲酯(純度99%)、丁香酚(純度99%)、丙烯醇(純度99%)、氯丙烯(純度99%)以及十四烷(純度99%),均購于阿拉丁化學試劑公司。第一代Hoveyda-Grubbs催化劑(C1)、第二代Grubbs催化劑(C2)、第二代Hoveyda-Grubbs催化劑(C3)和第一代Grubbs催化劑(C4)購自百靈威科技有限公司。油酸甲酯貯存于-18 ℃,催化劑和其他烯烴底物儲存于5 ℃。
ZNCL-GS智能加熱磁力攪拌器,重慶東俊儀器有限責任公司;SHZ-D(Ⅲ)型循環水式真空泵和RE-2000A型旋轉蒸發儀,鞏義市予華儀器有限責任公司;GC7090Plus氣相色譜儀,中國浙江福立公司;V3491雙排管,芯硅谷有限責任公司。
1.2 實驗方法
1.2.1烯烴復分解反應 反應通過雙排管在氮氣保護下進行,反應溶液均在手套箱中配制,稱取摩爾分數1%的催化劑(以原料油酸甲酯物質的量計,下同)至史萊克試管,隨后加入0.059 2 g(0.2 mmol)油酸甲酯、 2 mmol 指定烯烴底物和3 mL二氯甲烷,將試管從手套箱中取出連接至雙排管攪拌60 min,收集的產物使用帶有火焰電離檢測器(FID)的氣相色譜(GC)儀進行分析。
1.2.2GC條件 產物的氣相色譜(GC)分析在配備RB-5毛細管柱(30 m×0.25 mm×0.25 μm)的氣相色譜儀上進行,使用FID檢測器檢測組分。進樣口溫度250 ℃,檢測器溫度270 ℃。柱箱升溫程序如下:初始溫度60 ℃,保持5 min,以20 ℃/min升溫至220 ℃并保持10 min。分流比為30 ∶1,載氣為氮氣。用面積歸一化法與校正因子對產物及原料進行定量計算。
1.2.3校正因子及產率計算方法 通過查閱各物質的有效碳數表計算目標產物1-癸烯(CM1)、9-癸烯酸甲酯(CM2)與原料MO相對于內標物十四烷的校正因子(f),通過面積歸一化法得到較精確的產率和轉化率。
由氣相色譜儀分析結果計算得到MO轉化率和CM1、CM2產率,計算公式如下:
(1)
(2)
(3)
式中:C—MO的轉化率,%;Y1—CM1的產率,%;Y2—CM2的產率,%;S1—MO的峰面積;S2—CM1的峰面積;S3—CM2的峰面積;Si—內標物的峰面積;m0—初始加入的MO質量,g;mi—初始加入的十四烷質量,g;f1—MO的校正因子;f2—CM1的校正因子;f3—CM2的校正因子;M2—CM1的相對分子質量,140;M3—CM2的相對分子質量,170;n1—初始加入的MO的物質的量,mol。
2 結果與討論
2.1 油酸甲酯的自復分解反應篩選催化劑
當前典型的烯烴復分解反應催化劑有Schrock團隊開發的鉬基金屬卡賓催化劑,以及Grubbs團隊開發的釕基金屬卡賓催化劑,Schrock催化劑在產物的立體選擇性上有較好的表現,而Grubbs催化劑則具有更穩定的性能,可保持不被氧氣及水分破壞而實現對反應的高效催化[11-13],因此,Grubbs催化劑較適于本研究。通過4種市售可得的Grubbs催化劑(圖1)催化MO的烯烴自復分解(SM)反應,對該制備路線進行初探,自復分解反應流程如圖2所示,通過MO烯烴自復分解反應確認催化劑在本體系中的可行性,同時基于該反應達到平衡時的結果來探討底物結構對催化劑及反應路線的影響機制,反應結果如表1所示。
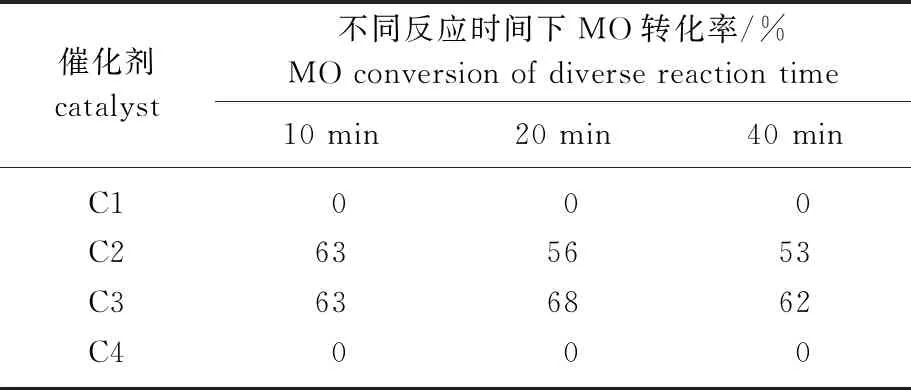
表1 MO自復分解催化劑篩選結果

圖1 釕金屬卡賓催化劑

圖2 自復分解反應流程
第一代Hoveyda-Grubbs催化劑(C1)與第一代Grubbs催化劑(C4)在該體系中沒有表現出活性,隨著反應時間延長,沒有觀察到MO的轉化,原因是MO的酯基與催化劑結合從而使其失活[14];第二代Grubbs
催化劑(C2)和第二代Hoveyda-Grubbs催化劑(C3)由于其中心釕金屬連有富電子大基團N,N雜環卡賓,保護催化劑不易受親核試劑進攻,可以穩定地催化MO進行烯烴復分解反應[15]。因此,在50 ℃、催化劑用量0.5%的條件下,催化劑C2和C3都在10 min 內催化MO轉化達到了63%的轉化率,其中,C2催化劑催化下的MO轉化率在20 min降至56%,在40 min時降至53%,而催化劑C3在相應時間分別變化至68%和62%,考慮C3更低廉的售價以及更高的MO轉化率,故以C3催化劑催化本反應,繼續考察底物對反應的影響機理。
2.2 底物對烯烴交叉復分解反應的影響研究

圖3 烯烴交叉復分解底物及反應流程
氧原子作為有孤對電子的親核試劑是進攻催化劑的主要因素,因此選取連有氧原子的烯丙基縮水甘油醚與2-甲氧基丙烯,同時選取連有芳香基團的苯乙烯和連有鹵素原子的氯丙烯作為底物進行考察,結果如表2所示。由結果可知,在原料與底物物質的量之比為1 ∶10、C3催化劑用量為1%、反應時間為40 min條件下,MO的轉化率隨溫度變化波動不大,而使用不同底物時獲得的MO轉化率差別較大。除氯丙烯外,其他底物下MO的轉化率隨溫度的變化幅度均不超過10個百分點。當苯乙烯為底物時,觀察到明顯的CM1和CM2的產率,在0 ℃時獲得了37%的CM1產率和35%的CM2產率。使用2-甲氧基丙烯作為底物時,沒有觀察到CM1和CM2產率,而MO的轉化率與自復分解時僅相差12個百分點。當底物為烯丙基縮水甘油醚時,MO的轉化率在50 ℃時為29%,低于自復分解的MO的轉化率,同時觀察到微量的CM1和CM2轉化率。由此可知,連有不同結構基團的短鏈烯烴底物對反應有較大影響,為了找到更合適本反應的底物,須對底物影響反應的原因作進一步分析。
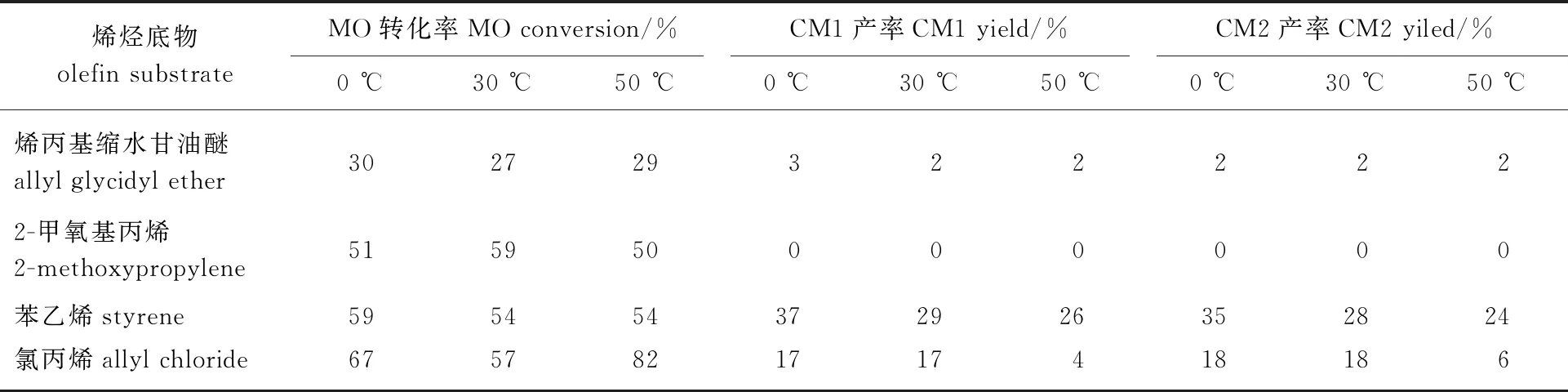
表2 烯烴底物對交叉復分解反應的影響
本研究基于目前公認的烯烴復分解Chauvin機理[19],以氯丙烯與苯乙烯為例,推測底物在催化反應中的具體影響方式,如圖4所示。催化劑C3的異丙氧基配體解離后活化,切斷一分子苯乙烯和氯丙烯分別形成中間體Ⅰ和Ⅴ,中間體Ⅰ和Ⅴ與原料MO絡合后發生[2+2]環加成反應形成釕金屬四元環烷基中間體Ⅱ和Ⅵ,經過開環后生成一分子產物和中間體Ⅲ,中間體Ⅲ再與MO絡合經加成成環與消除開環過程生成另一分子產物,完成一個催化循環。其中,在[2+2]生成四元環中間體時,由于底物所連基團不同,會造成其生成的難易程度不同,如中間體Ⅱ上的苯基與MO的長碳鏈互相排斥,導致此中間體較難生成,相應催化循環路線難以進行,因此無法獲得該催化路線所生成的目標產物,而中間體Ⅵ的氯原子與長碳鏈幾乎無排斥,中間體較易生成并順利完成催化循環,生成相應的α,ω-雙終端基團產物。
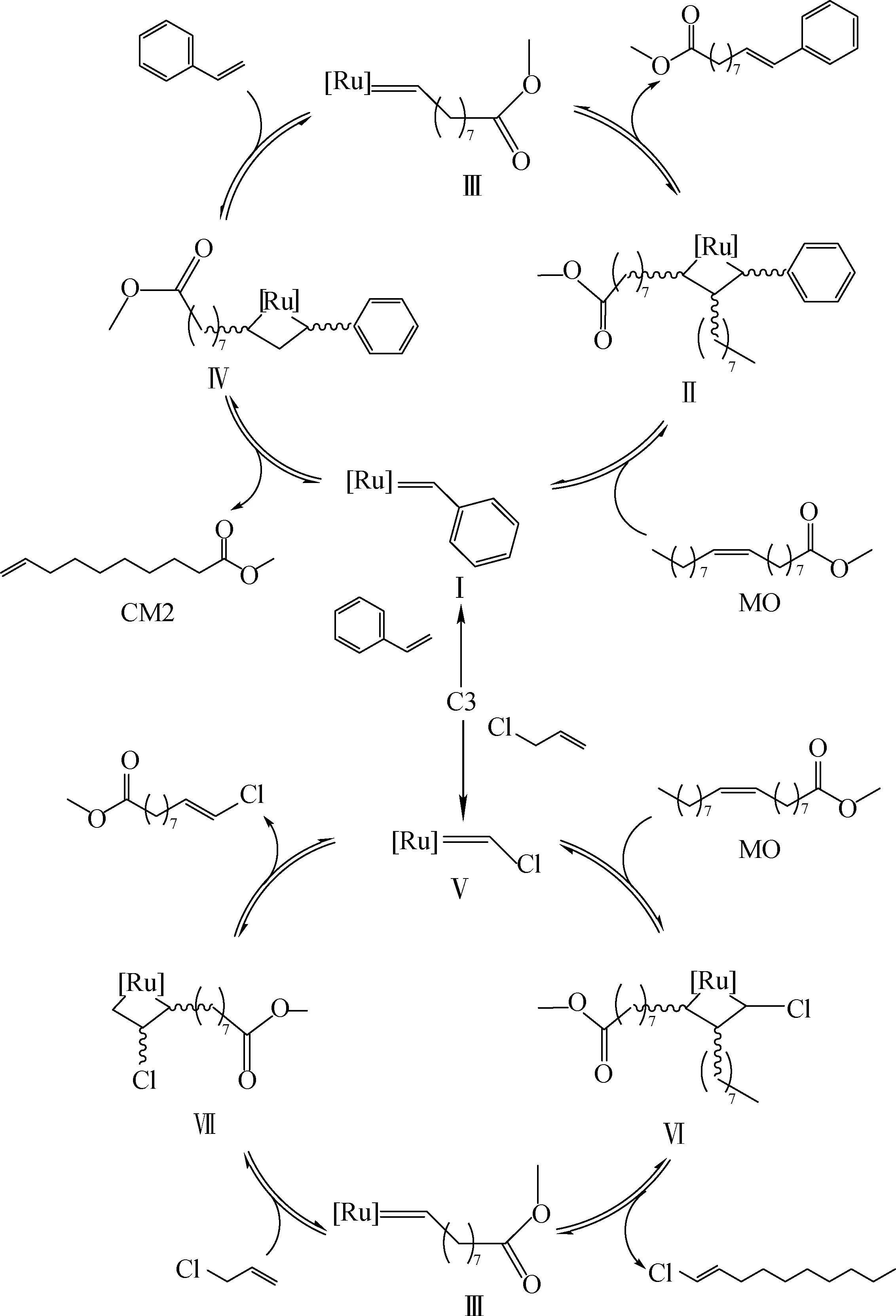
圖4 催化流程機理
由于苯環具有π鍵,能與雙鍵一樣進入過渡金屬的空軌道形成絡合,因此以苯乙烯為底物時,還可能有其他反應路徑,如圖5所示。催化劑配體解離后,釕金屬同時與苯環和雙鍵絡合形成螯合中間體Ⅷ,該中間體與MO進行[2+2]環加成反應形成四元環中間體Ⅸ,Ⅸ上苯環與金屬釕直接成鍵而遠離MO的長碳鏈,排斥現象不明顯,因此該中間體較易生成,Ⅸ在經歷消除反應生成產物CM1的同時形成中間體Ⅲ,Ⅲ與苯乙烯絡合進行[2+2]環加成反應形成中間體Ⅳ,由于MO雙鍵一端的長碳鏈在上一步四元環中間體Ⅸ的消除反應中隨產物CM1離去,于是在中間體Ⅳ上不存在與苯環排斥現象,因此該中間體的形成及其消除反應易發生,伴隨著產物CM2的生成,完成一個催化循環。

圖5 苯乙烯螯合機理
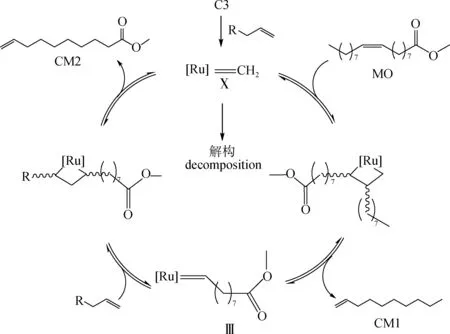
圖6 亞烷基金屬中間體流程
總之,苯環的大位阻令苯乙烯在催化劑與MO絡合形成的四元環中間體中與MO的長碳鏈存在排斥,不利于其參與的催化循環進行。而苯環和雙鍵與催化劑形成螯合中間體的排斥現象較弱,同時,該螯合中間體引導的催化循環可以同時生成終端烯烴化合物CM1和CM2。因此,連有苯環的苯乙烯是有利于本體系的底物。
2.3 驗證底物影響機理

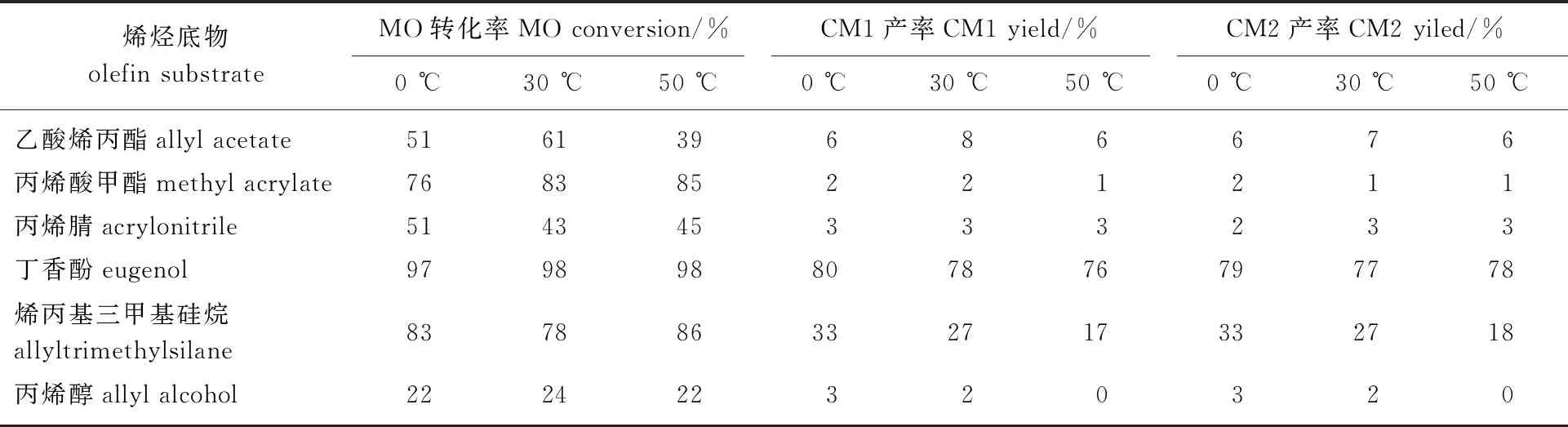
表3 不同底物交叉復分解反應結果
3 結 論
3.1通過烯烴復分解反應將油酸甲酯轉化為1-癸烯與9-癸烯酸甲酯,首先采用油酸甲酯的自復分解反應對烯烴復分解催化劑進行了篩選,結果表明:具有N-雜環卡賓配體的催化劑在本體系下的催化能力較好,油酸甲酯自復分解轉化率可達60%以上。然后選取連有不同類型基團的短鏈烯烴底物參與反應,發現底物對反應結果有較大影響,根據結果將其分為4種情況:一是不參與反應,代表底物是2-甲氧基丙烯;二是參與反應,但令催化劑失效進而反應終止,代表底物是烯丙基縮水甘油醚;三是獲取原料轉化率高,但反應路線不利于目標產物,代表底物是氯丙烯;四是有利于反應向目標產物的方向進行,代表底物是苯乙烯。
3.2基于烯烴復分解反應公認的Chauvin機理,結合對底物造成不同結果的劃分,進一步推測提出底物影響反應的具體機理,并選擇6種相似底物進行驗證,結果表明:連有大位阻和π鍵基團的烯烴底物在與催化劑絡合時能形成關鍵的螯合中間體,該關鍵中間體所參與的催化循環無明顯排斥現象,同時該催化循環所對應的產物是目標產物1-癸烯和9-癸烯酸甲酯。
猜你喜歡
大自然探索(2023年7期)2023-11-14 13:08:06
石油石化綠色低碳(2019年6期)2019-02-13 09:39:01
石油石化綠色低碳(2019年6期)2019-01-14 01:16:22
智富時代(2018年3期)2018-06-11 16:10:44
浙江大學學報(工學版)(2016年11期)2016-06-05 09:21:04
Coco薇(2016年2期)2016-03-22 02:45:06
超硬材料工程(2016年1期)2016-02-28 22:20:04
中國資源綜合利用(2016年4期)2016-01-22 08:27:23
合成化學(2015年4期)2016-01-17 09:01:27
應用化工(2014年3期)2014-08-16 13:23:50
