滇老鸛草藥材鑒別方法改進
2021-06-16 10:08:12黃云先趙健勝曹云蕓
中國民族民間醫藥 2021年9期
關鍵詞:方法
黃云先 趙健勝 曹云蕓
麗江市食品藥品檢驗所,云南 麗江 674100
滇老鸛草來源于牻牛兒苗科植物滇老鸛草GeraniumnepalenseSweet干燥地上部分。別名老鸛草、五葉草、老官草、六陽草、五瓣草、貓腳跡、老觀草[1]、尼泊爾老鸛草[2]。老鸛草來源于牻牛兒苗科的兩屬植物,即牻牛兒苗屬和老鸛草屬的不同植物[4]。牻牛兒苗屬植物我國約有四種,老鸛草屬植物我國約有55種,云南省約產21種。《云南省中藥飲片標準》2005年版第二冊收載的滇老鸛草為尼泊爾老鸛草GeraniumnepalenseSweet。老鸛草屬植物全草富含老鸛草鞣質,干葉含老鸛草鞣質、金絲桃苷,又含類沒食子酸、琥珀酸、鈣鹽、甜菜堿[3]、黃酮類槲皮素、山奈酚[2]等,《云南省中藥材標準》2005年版第二冊·彝族藥中對滇老鸛草的鑒別對照藥材薄層色譜鑒別,本實驗參照文獻[5],通過對云南省麗江古城區和玉龍縣5個批次的滇老鸛草樣品做了顯微鑒別及專屬性較強的成分沒食子酸的薄層色譜鑒別,以建立一個專屬性更強的鑒別方法,從而達到控制滇老鸛草藥材質量的目的。
1 儀器與實驗材料
1.1 儀器 德國塞多利斯BT224S電子分析天平;梅特勒MS205DU電子分析天平;Purelab Flex 超純水機。
1.2 實驗材料
1.2.1 試藥試劑 沒食子酸對照品(中國食品藥品檢定研究院,批號:110831-201906);老鸛草對照藥材(中國食品藥品檢定研究院,批號:121557-201102);三氯甲烷、甲酸乙酯 、甲酸、甲醇、、乙醇、乙酸乙酯環己烷、丙酮、正丁醇(分析純,天津市風船化學試劑科技有限公司);水(實驗室自制UP水,Ω=18.3); 硅膠GF254薄層板(青島海洋化工);硅膠G薄層板(青島海洋化工);硅膠G薄層板(美國莫克)。
1.2.2 樣品來源 采集麗江市玉龍縣、古城區不同五個地方的樣品共5個:
D1. 2015年9月,采收于麗江市玉龍縣白沙鄉玉湖村,為野生品種;為干燥地上部分。標本為全株(大樣)。
D2. 2015年6月,采收于麗江市古城區祥云街道辦事處吉祥社區,為野生品種;為干燥地上部分。
D3. 2015年9月,采收于麗江市玉龍縣黎明鄉黎明村,為野生品種;為干燥地上部分。標本為全株(溯源小樣)。
D4. 2015年7月,采收于麗江市玉龍縣拉市鄉,為野生品種;為干燥地上部分。
D5. 2015年5月,采收于古城區束河街道辦開文社區九子海,為野生品種;為干燥地上部分。
2 方法與結果
2.1 顯微鑒別(改進自擬方法) 本品莖橫切面 表皮由2~3列薄壁細胞組成,皮層為數列細胞,內側厚壁細胞環明顯,維管束8~9個,外韌型,呈輻射狀排列;形成層不明顯,韌皮部寬廣,木質部狹小,導管散列其中。薄壁細胞中草酸鈣簇晶眾多。
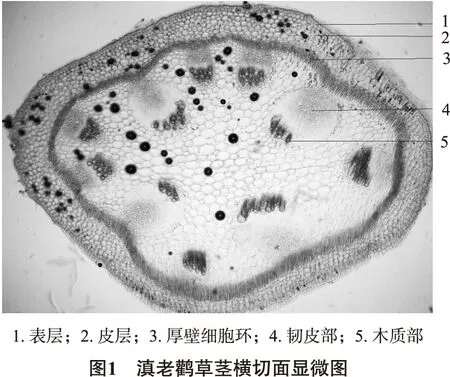
粉末顯微特征:本品粉末灰綠色。草酸鈣簇晶眾多而小,直徑10~20 μm。氣孔多為不定式,副衛細胞4~5個。非腺毛1~4細胞,基部細胞稍寬,尖端較長;腺毛短小,頭部圓形或橢圓形,腺柄1~4個細胞組成。螺紋導管多見;可見淀粉粒,多為單粒球形散在。見粉末顯微圖2A~F。

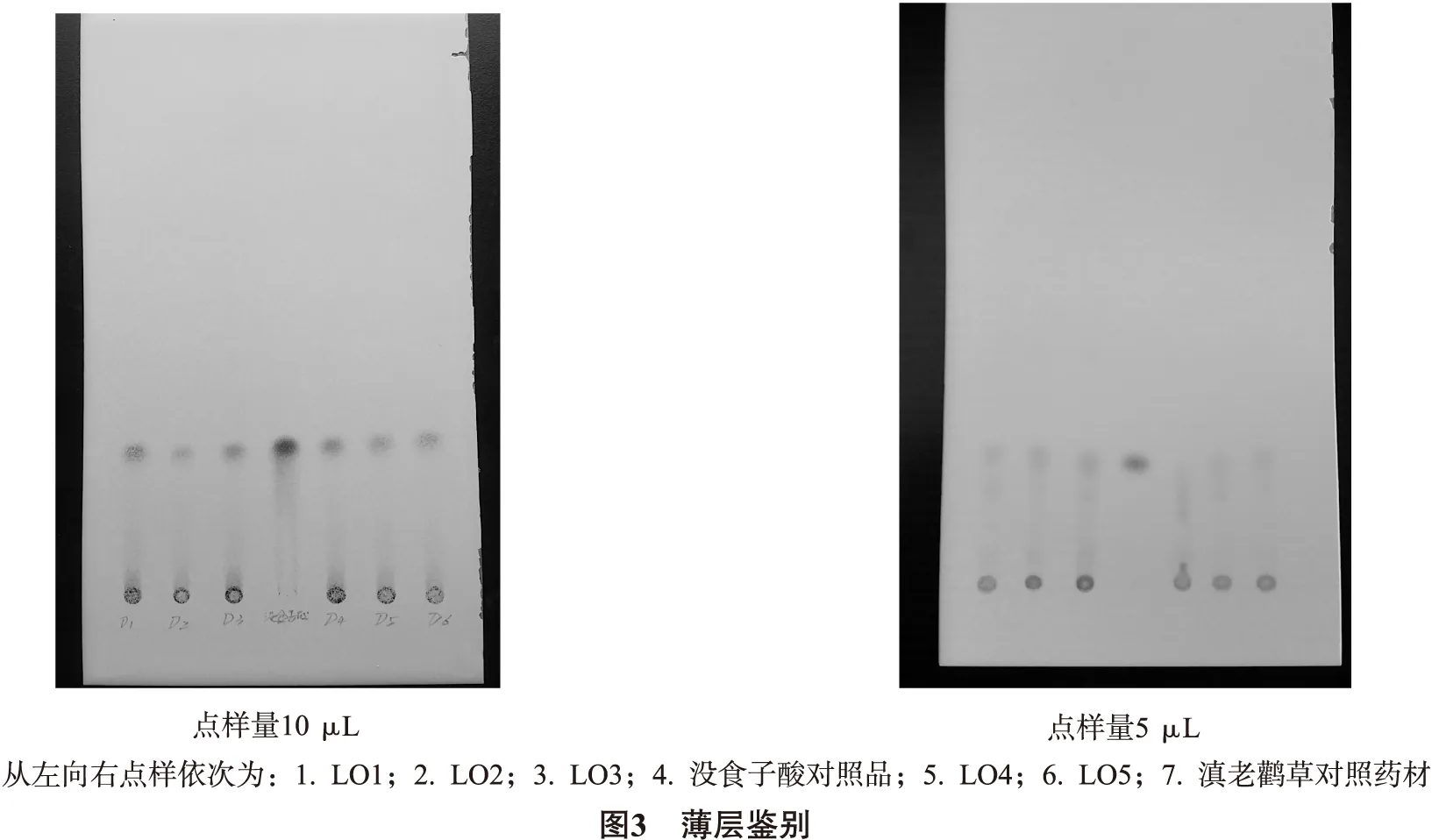
2.2 薄層鑒別(改進自擬方法) 取本品LO1到LO5號粉末(過四號篩)1 g,加水100 mL,加熱煎煮30 min,濾過,濾液用乙酸乙酯振搖提取三次,每次20 mL,取乙酸乙酯液水浴(60 ℃)上蒸干,殘渣加甲醇2 mL使溶解,作為供試品溶液。另取滇老鸛草對照藥材0.5 g,同法制成對照藥材溶液。取沒食子酸對照品,加甲醇制成每1 mL含1 mg的溶液,作為對照品溶液。照薄層色譜法《中國藥典》第四部附錄試驗,分別吸取上述兩種溶液各5 μL和10 μL,分別點于同一硅膠G薄層板上,以三氯甲烷-甲酸乙酯 -甲酸(5∶5∶0.8)為展開劑,展開,取出,晾干,噴以5%三氯化鐵乙醇溶液。供試品色譜中,在與對品色譜相應的位置上,顯相同藍色的斑點。不同點樣量的薄層圖如圖3所示。
3 分析方法驗證
3.1 樣品溶液制備試驗篩選 方法1:分別取本品粉末0.3 g和0.5 g(過四號篩),加甲醇20 mL,超聲30 min,放冷,濾過,濾液蒸干,殘渣加甲醇2 mL使溶解,作為供試品溶液。
方法2:分別取本品粉末0.3 g和0.5 g(過四號篩),加乙醇20 mL,超聲30 min,放冷,濾過,濾液蒸干,殘渣加甲醇2 mL使溶解,作為供試品溶液。
方法3:取本品粉末0.5 g,加70%甲醇20 mL,處理步驟同方法1。
方法4:取本品粉末0.5 g,加70%乙醇20 mL,處理步驟同方法1。
方法5:取本品粉末0.5 g,加50%甲醇20 mL,處理步驟同方法1。
方法6:取本品粉末0.5 g,加50%乙醇20 mL,處理步驟同方法1。
方法7:取本品粉末0.5 g,加30%甲醇20 mL,處理步驟同方法1。
方法8:取本品粉末0.5 g,加30%乙醇20 mL,處理步驟同方法1。
方法9:分別取本品粉末0.5 g和1 g,加水100 mL,加熱煎煮30 min,濾過,濾液用乙酸乙酯振搖提取3次,每次20 mL,取乙酸乙酯液蒸干,殘渣加甲醇2 mL使溶解。作為供試品溶液。
方法10:分別取本品粉末0.5 g和1 g,加水100 mL,加熱回流30 min,濾過,濾液用乙酸乙酯振搖提取3次,每次20 mL,取乙酸乙酯液蒸干,殘渣加甲醇2 mL使溶解。作為供試品溶液。
方法11:分別取本品粉末0.5 g和1 g,加水100 mL,加熱回流30 min,濾過,濾液用水飽和的正丁醇振搖提取3次,每次20 mL,取正丁醇液蒸干,殘渣加甲醇2 mL使溶解。作為供試品溶液。
方法12:分別取本品粉末0.5 g和1 g,加水100 mL,加熱煎煮30 min,濾過,濾液用水飽和的正丁醇振搖提取3次,每次20 mL,取正丁醇液蒸干,殘渣加甲醇2 mL使溶解。作為供試品溶液。
通過實驗室實驗,發現方法9操作可行性高,分離效果良好,方法驗證效果良好,決定選用方法9中取1 g的方法為最終供試品制備方法。
3.2 展開劑篩選 展開劑1:乙酸乙酯-甲醇 -水(8∶5∶0.2);
展開劑2:乙酸乙酯-甲醇 -水(8∶5∶2)的下層溶液;
展開劑3:三氯甲烷-甲醇-水(8∶5∶0.2);
展開劑4:環己烷-乙酸乙酯-甲醇(6∶2.5∶1);
展開劑5:三氯甲烷-甲酸乙酯-甲酸(5∶4∶1);
展開劑6:三氯甲烷-甲酸乙酯-甲酸(5∶5∶0.8);
展開劑7:甲苯(水飽和)-甲酸乙酯-甲酸(10∶8∶1);
展開劑8:乙酸乙酯-丁酮-甲酸-水(5∶3∶1∶1);
展開劑9:正己烷-甲醇(1∶ 9) ;
展開劑10:正己烷-乙酸乙酯(1∶4)。
飽和10 min,上行展開。
通過比對用展開劑6號展開時RF值為0.37,在0.2~0.8藥典規定的范圍,展開效果好。
3.3 檢視方法的選擇 噴以10%磷鉬酸顯色;噴以2%三氯化鐵50%的乙醇溶液;噴以5%氯化鐵乙醇液;紫外光燈(365 nm)下檢視;噴以5%三氯化鐵乙醇液,再在紫外光燈(365 nm)下檢視。選用上述5種方法檢視,第三種斑點清晰且方法簡便。
3.4 點樣方式及點樣量考察 點樣方式為接觸點樣;點樣量為2 μL、4 μL、5 μL、6 μL、8 μL、10 μL。溫度及相對濕度的影響 按照試驗所得薄層色譜條件,考察本品在溫度5~30 ℃、相對濕度10%~75%展開時,對薄層色譜結果的影響。結果表明,在上述溫度、濕度范圍內對該薄層色譜結果無明顯影響,點樣量為10 μL斑點效果好。
3.5 預平衡試驗 按照試驗所得薄層色譜條件,考察未平衡、平衡15 min、平衡30 min展開,對薄層色譜結果的影響。結果表明,未平衡的展開邊緣效應較明顯,平衡15 min與平衡30 min展開,所得分離度均較好,且無明顯差異。因此采用預平衡15 min展開。
3.6 重復性 不同產地的5批次樣品的檢測結果表明,該方法具有重復性。
4 討論
4.1 顯微鑒別方法建立 顯微鑒定作為鑒別中藥、中成藥的手段之一,其方法可變性不強、操作簡便、而特異性較強,可以說利用顯微特征對比鑒定對大多數中藥材都行之有效。經實驗,滇老鸛草也不例外,的確具有其顯著的特異性。
4.2 引進代表性對照品 民族藥標準現階段整體質量標準水平較低,仍多集中于藥材基源及性狀鑒定的研究,尚未建立指標性成分的質量控制,原標準未引進代表性對照品進行定性。通過大量查找資料,考慮滇老鸛草的傳統主要功效為止瀉止血,根據文獻報道,老鸛草屬植物主要含鞣質類沒食子酸、黃酮類槲皮素、山奈酚等,故考慮引進沒食子酸作為其代表性成分研究制定薄層色譜鑒別方法。
4.3 薄層鑒別方法建立 依據沒食子酸易溶于水、醇和醚;具有酚(易被氧化和三氯化鐵水溶液生成藍黑色沉淀)及羧酸(加熱時失去二氧化碳成焦性沒食子酸)的性質。試驗考察了采用不同取樣量,選用水及不同比例的甲醇和乙醇采用超聲、加熱煎煮及回流提取方法,跟著考察了水提的提取液再用不同的有機溶劑乙酸乙酯、正丁醇及水飽和正丁醇提取的多種方法做對比,最終實驗驗證采用方法9提取效果較好,能有效的提出沒食子酸。
展開劑的選擇在使用不引入甲酸的展開劑如1~4號及9號10號展開時,有的在原點形成堆積或展開不上去,用7號及8號展開劑展開,結果斑點不太清晰,5號和6號展開劑的選擇參考了《中國藥典》2015版中五倍子中鞣質類成分沒食子酸薄層鑒別方法中的三氯甲烷-甲酸乙酯-甲酸(5∶5∶1,V/V/V)。實驗考察了展開劑中甲酸比例對展開效果的影響,結果是6號展開劑三氯甲烷-甲酸乙酯-甲酸(5∶5∶0.8,V/V/V)展開能使沒食子酸處于游離狀態,斑點分離效果好,Rf值適中,且無拖尾現象。
最終采用方法9制備供試品溶液,硅膠G薄層板,點樣量10 μL,以展開劑6號15 mL為展開劑,展開,取出,晾干,噴以5%三氯化鐵乙醇液,所得薄層色譜斑點清晰,分離較好,易判斷結果,且重現性好。
5 結論
此次實驗新擬的顯微鑒別方法操作簡便、特異性強,建議標準修訂時增加顯微鑒別。薄層色譜條件可行有效,原標準中鑒別項下未引進代表性對照品進行定性,考慮引進沒食子酸作為其代表性成分,建議標準修訂時增加現擬方法,應與沒食子酸對照品及滇老鸛草對照藥材色譜相同。可見以沒食子酸作為滇老鸛草藥材標識性成分有其合理性。可用于滇老鸛草藥材的質量控制,以提高民族藥材的質量標準,也可用于制定滇老鸛草對照藥材標準研制的起草。
猜你喜歡
中老年保健(2021年9期)2021-08-24 03:52:04
河北畫報(2021年2期)2021-05-25 02:07:46
中學生數理化(高中版.高考理化)(2020年2期)2020-04-21 05:33:04
兒童繪本(2020年5期)2020-04-07 17:46:30
兒童故事畫報(2019年5期)2019-05-26 14:26:14
Coco薇(2016年2期)2016-03-22 02:42:52
山東青年(2016年1期)2016-02-28 14:25:23
Coco薇(2015年1期)2015-08-13 02:47:34
小雪花·成長指南(2015年7期)2015-08-11 15:03:12
小雪花·成長指南(2015年4期)2015-05-19 14:47:56
