mTOR信號通路在糖尿病腎病發病機制中的研究進展
2021-06-02 09:01:56王珍楊洛廖敏郝亞榮
生物技術進展 2021年3期
王珍, 楊洛, 廖敏, 郝亞榮
武漢大學人民醫院老年病科,武漢 430060
糖尿病是全球導致死亡的主要疾病之一,是重大公共衛生問題。世界衛生組織統計大約有4.22億人患糖尿病,而且患有糖尿病的人數呈逐年上升趨勢。研究表明,1/5的糖尿病患者可能有腎病并發癥[1]。糖尿病腎病(diabetic nephropathy,DN)已經成為糖尿病最常見的并發癥。由于DN的高發病率以及與晚期腎病、心血管疾病和過早死亡等風險相關,其已成為全球性的健康問題[2-3]。當前的糖尿病患者的管理重點是嚴格控制血糖、降壓和降脂,然而這些干預不能在很大程度上降低DN患者的比例[4-5]。鑒于DN治療方案的局限性,研究人員一直在努力闡明腎損害的信號通路,從而開發新的藥物。DN是一種多因素的疾病,其特征是復雜的相互作用的血流動力學和代謝因子,包括高血糖水平、晚期糖基化終產物和腎素-血管緊張素-醛固酮系統的激活[6]。因此,DN的發病機制涉及多種信號通路。在正常血糖水平下,自噬是腎上皮細胞(包括足細胞、近端腎小管、系膜和內皮細胞)的重要保護機制,但是在高血糖狀態下,自噬的活性下調,可導致DN的發生和進展;慢性的全身性炎癥和炎癥反應,如循環細胞因子的增加,也已被認為是DN發展的主要因素[7];DN還與氧化應激增加以及碳水化合物、脂類和蛋白質代謝的改變有關。
雷帕霉素靶蛋白(mammalian target of rapamycin,mTOR)是雷帕霉素的靶蛋白,是一種絲氨酸/蘇氨酸蛋白酶,是磷脂酰肌醇3激酶相關激酶蛋白家族的成員。mTOR主要參與磷酸肌醇3-激酶(phosphoinositide-3-kinase,PI3K)/蛋白激酶B(protein kinase B,Akt)/mTOR信號通路,在延遲細胞凋亡以及促進細胞分裂、細胞存活和血管生成中發揮了主導作用[8]。mTOR包含2種不同的蛋白復合物,分別為雷帕霉素靶蛋白復合物1(mechanistic target of rapamycin complex 1,mTORC1)和mTORC2,它們可以通過其不同的底物來區分。mTORC1基本的核心物質包括mTOR、G蛋白β亞基、mTOR的雷帕霉素敏感接頭蛋白和分子量為40 kDa的含脯氨酸Akt底物。mTORC1的主要作用為促進蛋白質合成和細胞體積的增加;其還可以感知并響應細胞內外營養水平的波動,主要是生長因子、細胞能量、氧水平、氨基酸等;活化的mTORC1可導致2個下游靶點核糖體S6激酶和真核翻譯起始因子4E結合蛋白磷酸化,這2個靶點刺激核糖體合成和蛋白質翻譯,從而誘導細胞的生長和增殖,以增加細胞質量[9]。mTORC2基本的核心物質包括mTOR、哺乳動物應激激活的蛋白激酶反應蛋白、G蛋白β亞基和Rictor蛋白,其主要作用是控制細胞存活和構建細胞骨架[10]。
近年來,多項研究致力于探索mTOR在DN發病機制中的作用,結果表明,在DN的動物模型和患者中,mTOR在多條信號通路中位于核心位置,參與了腎臟多種病理過程,如自噬、炎癥、氧化應激等[11-12]。因此,本文就mTOR介導的自噬、炎癥及氧化應激信號通路在DN發病機制中的相關研究進展做一綜述,以期為DN治療及預防提供理論參考。
1 自噬
自噬是一種高度保守的分解代謝過程,是一種細胞自我保護機制。在缺氧等條件下,自噬被激活,其主要通過溶酶體和雙膜自噬體的整合識別錯誤折疊的蛋白質、受損細胞器、有害的胞內物質并將這些物質降解為可利用的產物,通過再循環為細胞提供能量用于修復和再生,以維持腎小球和腎小管的穩態。在高糖條件下,自噬水平降低,細胞內的細胞器和大分子積累且無法清除,從而導致腎臟足細胞、腎小球、腎小管損傷,最終可出現DN的發生和發展[13-14]。可見DN與自噬活性的減低明顯相關。自噬的作用機制受多種因子調控,其中主要的負調控因子是mTOR[15]。因此,通過抑制mTOR激活來調節自噬的活性可能是DN的重要治療靶點[16]。
1.1 mTOR介導自噬上游信號通路
在腎臟足細胞自噬過程中,信號傳遞、自噬運動和囊泡融合都涉及PI3K/Akt途徑。PI3K是真核生物中調節自噬的重要信號分子,Akt是PI3K/Akt信號通路的核心[17]。研究表明,DN模型組會出現腎小球數量、形態改變,腎臟微管相關蛋白1輕鏈(microtubuler associated protein 1 light chain,LC)和Bcl-2相互作用蛋白(Bcl-2 interacting coiled-coil protein 1,Beclin-1)表達顯著降低,二者的含量與自噬呈正相關,是評估自噬活性水平的最佳標志[18],而PI3K、Akt和mTOR表達顯著升高。這表明DN小鼠通過PI3K/Akt/mTOR通路的激活,抑制了腎臟自噬保護機制,導致腎臟病理改變[19]。
1.2 mTOR介導自噬下游信號通路
自噬途徑主要由自噬相關基因(autophagy-relatedgene, Atg)蛋白控制,Atg蛋白可參與組成ULK1(UNC-51-like kinase 1)復合物和液泡分選蛋白34(vacuolar protein sorting 34, Vps34)復合物。ULK1是mTORC1信號傳導至自噬細胞的關鍵介質。當上游信號通路傳導活化后,mTOR被激活,ULK1中的mTORC1依賴性的磷酸化位點發生去磷酸化。ULK1可抑制分解代謝過程,從而導致自噬受到抑制。自噬誘導所需的另一種自噬復合物是Vps34復合物。活化的mTORC1使Vps34復合物磷酸化,進而抑制自噬體的合成并抑制自噬[17]。
1.3 其他信號分子影響mTOR活性介導自噬
近年來,多項研究表明各種信號分子可通過影響mTOR活性介導自噬。維生素D通過下調mTOR基因表達、阻斷mTOR通路可降低鏈脲菌素誘導的糖尿病腎小球平滑肌肌動蛋白α的表達、系膜基質積累和腎臟肥大,增強自噬作用[20]。KCa3.1鉀通道通過激活PI3K/Akt/mTOR信號通路介導DN患者的自噬活性降低[18]。高血糖誘導的硫氧還蛋白互作蛋白通過激活mTOR信號通路參與糖尿病腎病小管自噬和線粒體自噬的異常調節(圖1)[21]。此外,青錢柳富含的三萜酸可顯著增加AMPK的磷酸化,并降低其下游效應物mTOR的磷酸化,從而增加自噬活性,以延緩DN的發展[22]。
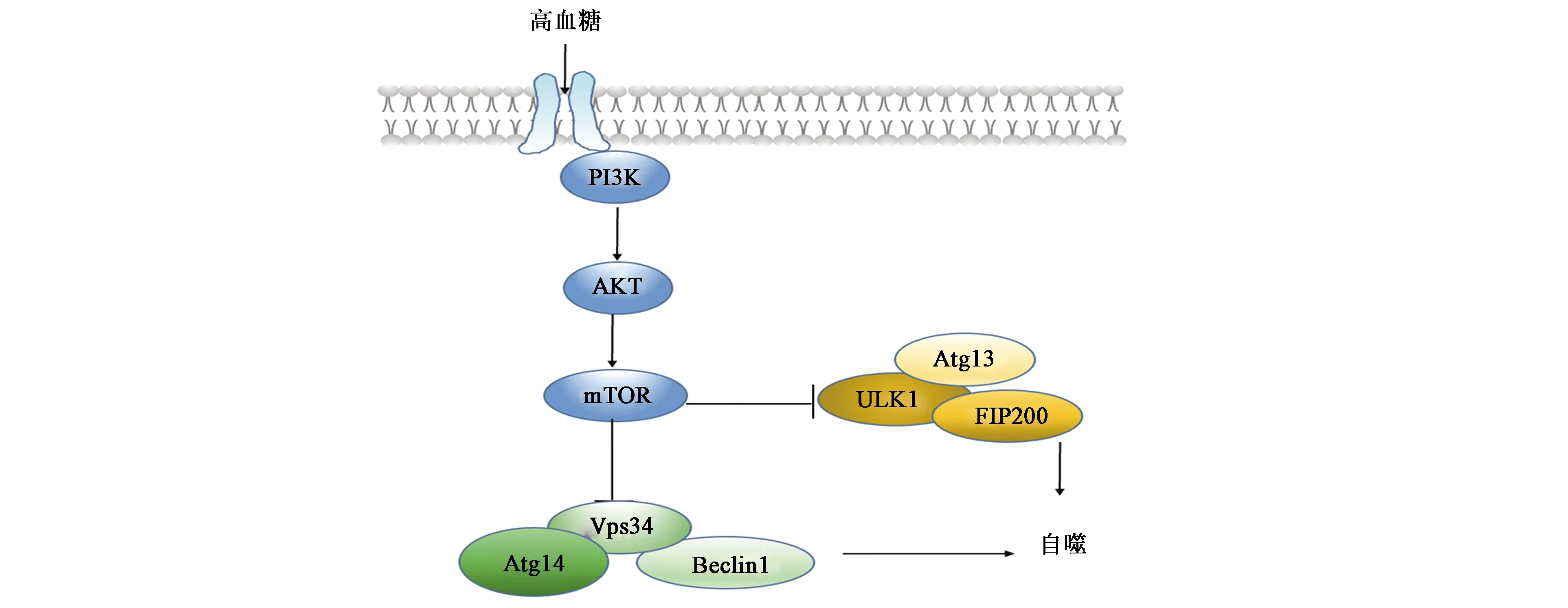
圖1 mTOR介導的自噬信號通路
2 炎癥
炎癥是機體對有害條件作出反應時激活的一種機制,以維持組織的穩態和完整性。炎癥反應的慢性激活會引發許多副作用。己知高血糖、炎癥和腎素-血管緊張素-醛固酮系統激活是糖尿病相關腎損害的始動因素,并且大量證據表明炎癥在糖尿病并發癥的發生和發展中起著關鍵作用[23]。在2型糖尿病(type 2 diabetes mellitus,T2DM)患者中,促炎細胞因子如白細胞介素-1β(interleukin-1β, IL-1β)、白細胞介素-6、腫瘤壞死因子α和C-反應蛋白(C-reactive protein, CRP)的水平升高,提示炎癥與T2DM有密切關系[23-25]。DN介導的腎損傷中的炎癥反應,可以通過抗炎治療,以預防和延緩腎臟損害的進展[25-27]。因此,在過去的幾年中,炎癥反應的調節已成為降低DN發病率的一種可能的治療方法。高血糖是一種糖尿病及其并發癥主要的上游機制,而炎癥被認為是糖尿病并發癥進展的下行驅動力[28]。多種炎癥因子可以通過mTOR介導的信號通路介導腎臟炎癥反應,使腎臟結構發生破壞。
IL-1是一種常見的炎性因子。研究表明IL-1可顯著影響腎臟功能,IL-1與DN發生和發展關系密切,而且IL-1含量增高會促進DN的發生和發展。研究表明,IL-1可直接結合PI3K的調節性p85亞單位[29];在動物實驗模型中加入IL-1后,PI3K/Akt/mTOR途徑可迅速激活[30-32]。總之,IL-1可以激活PI3K/Akt/mTOR信號通路,導致腎臟發生炎癥反應。
CRP是一種急性期蛋白,在炎癥和組織損傷時迅速合成和釋放。在T2DM患者中,CRP水平升高與微量白蛋白尿和腎功能不全密切相關,提示CRP水平很有可能用來衡量DN炎癥的嚴重程度[33]。CRP可以激活人腎小管上皮細胞mTOR的信號機制,從芯片分析可以清楚地檢測到果蠅母性DPP同源蛋白3(sekclsky mothers against dpp3,Smad3)與mTOR的非翻譯區(untranslated regions, UTR)區域的物理互作,CRP在很大程度上增強了這種結合位點。而定量流式細胞術、免疫熒光和Western blot分析的結果進一步證明了Smad3和mTOR信號之間的功能聯系,即用特定抑制劑阻斷Smad3信號能夠抑制CRP誘導的mTOR信號,揭示了CRP與Smad3及mTOR信號有相互作用關系[34]。在高糖條件下,小鼠喂養時添加CRP或高糖能夠激活mTOR,CRP和高糖的聯合應用并沒有增加mTOR信號;然而,添加中和抗體CD32b可減弱高糖和CRP誘導的mTOR信號,這意味著高糖通過CRP-CD32b機制誘導mTOR信號[34]。總之,CRP可通CD32b/Smad3/mTOR促進腎臟炎癥信號傳導機制,證明mTOR信號通路是CRP促進2型糖尿病腎病的一個中心機制。
3 氧化應激
氧化應激是DN發病的一個主要的病理生理學事件。在正常的生理條件下,氧化與抗氧化系統處于動態平衡狀態,使生物體免受活性氧的毒性。然而,在不同的病理生理條件下(如缺血再灌注、慢性腎功能衰竭和DN等),腎小球細胞的氧化應激成為腎臟損害的主要原因[35]。高血糖是DN的主要發病因素。高血糖抑制腎實質細胞對于組織葡萄糖的攝取,導致機體有氧氧化水平降低,抑制線粒體氧化呼吸鏈,使代謝產物積累,包括H2O2等物質,即增加了活性氧的產生[36-37]。因此,高血糖是DN的主要致病因素之一,并且自氧化、非酶化蛋白糖基化和增加多元醇途徑都與血糖升高有關。這些機制引發氧化應激,并可能影響許多細胞代謝活動。研究表明抑制mTOR可以改善高糖誘導的腎臟細胞的氧化應激狀態[38]。
3.1 mTOR介導氧化應激的信號通路
高糖誘導的活性氧(reactive oxygen species,ROS)過度表達是糖尿病并發癥的統一機制。并且,高血糖狀態下ROS的過度生成在糖尿病并發癥的發病機制中起著核心作用,線粒體源性ROS是引起細胞死亡和腎功能不全的主要細胞內活性氧來源[39-41]。而ROS可調節Akt/mTOR信號傳導。相關實驗發現,雷帕霉素可抑制mTOR,降低高糖暴露腎小球系膜細胞的ROS生成,提示mTOR參與高血糖狀態下ROS的生成[42]。在另一實驗中,結果同樣證實了mTOR介導了DN的氧化應激。mTOR的抑制可以顯著降低體內外還原型煙酰胺腺嘌呤二核苷酸磷酸(nicotinamide adenine dinucleotide phosphate,NADPH)氧化酶的活性。NADPH氧化酶4是NADPH氧化酶復合物的成員,可以產生ROS并在介導線粒體功能障礙和細胞凋亡中發揮重要作用。mTOR抑制可降低NADPH氧化酶水平[43]。這些結果表明mTOR通過激活NADPH氧化酶增加ROS的產生。另一方面,在高糖條件下,超氧化物歧化酶活性明顯降低。相反,在糖尿病條件下,mTOR通路的阻斷導致谷胱甘肽水平和超氧化物歧化酶活性升高,這表明mTOR抑制劑增強了清除有害氧化的能力[44]。因此,mTOR抑制劑可通過上調抗氧化酶和下調NADPH氧化酶活性來保護腎臟免受氧化應激的影響。
3.2 其他藥物影響mTOR介導氧化應激
多種mTOR抑制劑同樣可通過上調抗氧化酶和下調NADPH氧化酶活性來保護腎臟免受氧化應激的影響。抗菌肽-BF(BF-30)有抗微生物和抗炎作用[44]。實驗結果顯示,BF-30可以增加人腎小管上皮細胞單磷酸腺苷激活的蛋白激酶(adenosine 5′-monophosphate-activated protein kinase, AMPK)的磷酸化,抑制mTOR的磷酸化,并促進LC3的降解,提示其能有效拮抗氫過氧化物誘導的凋亡,抑制腎組織氧化應激,減少腎臟纖維化,增加自噬和減少腎細胞凋亡相關蛋白,減少細胞損傷,保護腎細胞[45]。三七素也具有同樣的作用。研究表明,在腎小管內皮細胞中三七素實驗組的活性氧減少,胞內抗氧化酶增加,同時mTOR的活性降低,提示三七素可以通過抑制mTOR的活性,改善氧化應激,從而保護糖尿病大鼠的腎組織[46]。巖白菜素能抑制葡萄糖誘導的腎小球細胞外基質的產生,通過mTOR/轉錄重復包含蛋白1/核轉錄相關因子2途徑下調系膜細胞的氧化應激,因此巖白菜素可能成為預防和治療DN的候選藥物[47]。
4 展望
DN是慢性腎臟病的主要病因之一,也是糖尿病患者死亡的主要原因,但對糖尿病引起的腎損傷的潛在機制知之甚少。DN患者現有的治療方法包括控制血糖、膽固醇和高血壓等,可并不能明顯減低DN患者比例以及延緩DN的進展。因此,亟需闡明糖尿病腎損害的信號通路,以開發新的藥物。激活mTOR可以抑制自噬、促進炎癥、促進氧化應激,導致DN的發生和發展。了解了mTOR信號通路在DN發病過程中的作用,對未來DN治療策略具有重要意義。這為臨床治療DN提供了一種新思路。
然而,目前關于mTOR信號通路在DN發病機制中的研究進展多是關于mTOR介導自噬的信號通路,關于炎癥及氧化應激的研究很少。且mTOR信號通路的研究尚未完全透徹,尤其關于mTORC2的研究較少。因此,還應該多關注mTOR介導DN炎癥及氧化應激的信號通路及mTORC2介導DN的信號通路。隨著更多研究中mTOR信號通路在DN中機制的進一步闡明,DN的預防及治療必將更進一步。
猜你喜歡
中老年保健(2022年5期)2022-08-24 02:35:42
中老年保健(2022年1期)2022-08-17 06:14:56
中老年保健(2021年5期)2021-08-24 07:07:20
中老年保健(2021年11期)2021-08-22 03:15:16
世界科學技術-中醫藥現代化(2020年2期)2020-07-25 02:05:56
文苑(2018年21期)2018-11-09 01:23:06
西南軍醫(2016年6期)2016-01-23 02:21:19
中國衛生(2015年9期)2015-11-10 03:11:12
西南軍醫(2015年2期)2015-01-22 09:09:37
中國衛生(2014年3期)2014-11-12 13:18:12
