基于深度學習的染色質交互作用預測
2021-05-13 07:16:04任景瑞李川張振毓鄧凱
現代計算機 2021年8期
關鍵詞:結構
任景瑞,李川,張振毓,鄧凱
(1.四川大學計算機學院,四川610065;2.四川大學網絡空間安全學院,四川610065)
0 引言
染色質是由DNA、組蛋白、非組蛋白等多種物質組成的遺傳物質,其結構復雜,難以直接觀察,但對細胞遺傳過程的基因表達有重要影響。自3C 技術問世以來,眾多方法被陸續報道用于捕獲染色質構象,其中Hi-C 技術是捕獲染色質相互作用頻次的最新最常用方法[1]。Hi-C 原始互作數據可以通過交互頻次的讀取序列映射到對稱矩陣中,并且利用這種矩陣熱圖可以表示并構造為染色質的高級結構TAD[2]、隔間和染色質環等。染色質的高級結構與其功能密切相關,對基因表達和生物遺傳有重要影響[3],如三維結構變化可能誘導腫瘤發育產生[4]。
目前對染色質結構的研究非常豐富,由于Hi-C 數據測量非常昂貴且耗時,但對與基因表達、轉錄和疾病狀態相關的染色質結構有重要意義[5],所以有很多研究開始關注減少實驗進行預測,對染色質結構研究存在三維結構構建,二維結構預測以及基因表達和功能性研究。對染色質三維構建目前存在多種方法,分別使用了多種距離模型算法來構建三維空間結構[6]。最新的三維結構構建方法還可以分析其基因功能[7]。而二維結構分析同樣基于Hi-C 數據區域分割[8]或DNA 序列預測[9]來構建TAD、染色質環等結構,識別其區域性。染色質測序技術的發展,還對表觀遺傳學方面的實驗應用非常重要[10],表觀遺傳學包括組蛋白修飾等方面,對基因表達調控和染色質重塑有重要影響[11]。分析組蛋白修飾的功能作用[12],研究染色質結構功能和基因表達[13]的影響,例如對染色質開放性[14]和染色質狀態的識別[15],具有重要研究價值。
利用深度學習預測染色質交互作用能夠有效獲取輸入數據的前后關聯信息和局部特征,目前對人類細胞類型GM12878[16]和果蠅細胞類型[17]都有相關報道,使用的數據一般圍繞DNA 序列和表觀遺傳學數據。為了深入研究組蛋白修飾和染色質交互作用兩種數據相關性,本文提出了一種從組蛋白修飾數據中預測人類基因組中Hi-C 數據的方法,基于卷積神經網絡,針對常見人類細胞類型IMR90 建立了深度學習模型比較預測,通過線性相關系數皮爾遜系數以及圖相似性系數等評估,并最終在預測結果與原始結果之間表現出高相關性。
1 數據和方法
1.1 數據預處理
Hi-C 數據:在GEO 公開數據集上,可以通過訪問代碼GSE63525 下載IMR90 細胞類型的數據。我們從實驗原始觀測的Hi-C 序列交互數據生成原始矩陣數據,并根據分辨率確定每段基因的長度,計算對應段位置(例如本文采用10kb 分辨率,k 為一千數量,b 指代堿基段即一段堿基序列,就會將每個交互位置數據除以104,獲得其bin 序號,每條染色體按細胞類型和染色體不同有上百萬或上億堿基長度)。本文關注研究染色質內交互作用,即同一序號染色體間的交互作用(同染色質間交互),最終獲得22 條染色體的Hi-C 原始交互作用熱圖(去除性染色體影響)。
組蛋白修飾數據:從Roadmap 上可以下載每種細胞類型的所有表觀遺傳修飾數據,https://egg2.wustl.edu/roadmap/data/byFileType/signal/consolidated/macs2signal/foldChange/。
不同的細胞格類型對應有不同的組蛋白修飾。對于IMR90 細胞類型,可以下載以下多種修飾因素:
H3K23ac、H3K79Me1、H3K27Ac、H3K79me2、H3K27me2、H3K79me3、H3K27me3、H3K9acH3K3K36me1、H3K9me1、HP4、RPD3、H1、H3K36me2、H3K36me3、H3K9me3、H3K-4me1、H4、H3、H3、3、3me1、H4K3、3k4me1、H4K16ac
以上數據可在ENCODE 項目中公開訪問,首先對組蛋白修飾的數據進行預處理,保持與Hi-C 數據同樣分辨率大小(例如大小為10kb)。
1.2 實驗環境
GPU:NVIDIA TU102[GeForce RTX 2080 Ti Rev.A](rev a1)
CPU:48 英特爾至強CPU E5-2650 v4@2.20GHz
內存:128GB
Python 版本和依賴包環境:Python 3.6,基于TensorFlow 的Keras。
1.3 模型與評估
本文通過多種的深度學習方法評估訓練結果。訓練神經網絡并使其擬合的過程重,使用MSE(Mean Square Error)作為損失函數,使用MAE(Mean Absolute Error)作為目標函數。
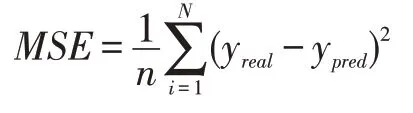
線性相關性上使用皮爾遜相關系數(PCC)和斯皮爾曼相關系數(SPCC)來評估預測結果,結果數值將顯示預測結果與原始數據之間的線性相關性,相關系數的絕對值越接近1,其相關性就越強。
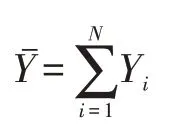
皮爾遜相關系數公式:
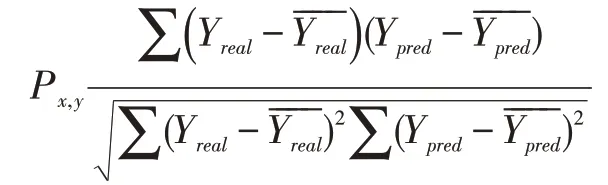
SPCC 是基于PCC 的一種相關系數計算方法,給定變量基礎上給出一個排序差異集合d,由兩個變量集合中的每個元素計算,最后使用PCC 公式來獲取排序變量的結果。
除序列評估外,還可使用計算峰值信噪比(Peak Signal to Noise Ratio)和結構相似度指數(Structural SIMilarity)來進行評估,這兩者都常用于圖像處理和去噪。
PSNR 表示圖像信噪比,此值越大代表失真越少,MAX=max{Ypred}-min{Ypred}。
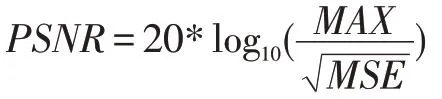
SSIM 值的范圍從0 到1,衡量兩幅圖的相似度,判斷預測圖像是否接近于原始圖像:
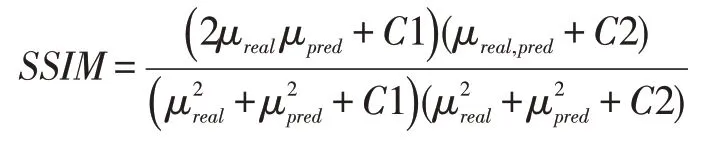
2 方法
2.1 數據預處理
對于輸入數據和輸出數據,本文分別使用不同方法進行預處理,因為不同數據實驗測序方法不同,其表達值彼此差異較大,難以直接定性分析。
Hi-C 數據可使用標準化函數將其歸一化到[0,1]的區間范圍,表示每個位置交互的可能性,其原始序列交互數據可從Rao 等人(GSE63525)[18]所做的公開可用的Hi-C 實驗中獲得。原始數據為每個染色質每段堿基部分間的交互作用強度,在10k 堿基分辨率下,每個作用強度數據就表示染色質上按順序排列的兩段10k 長度堿基的交互頻率。由于每個染色質長度彼此不同,使用字母標識i 和j 表示兩個段堿基序號,它們之間的計數nij 表示染色質段上相互作用次數,通過堿基位置對應關系可構成對稱矩陣,矩陣大小為N 表示染色質長度L/分辨率R(如圖1 所示)。
對于組蛋白數據,首先通過bwtools 和指定bin 長度(分辨率)生成組蛋白修飾序列信號數據矩陣H,此分辨率與Hi-C 數據分辨率一致,且進行截取使實驗數據長度相同,矩陣H 中每列為組蛋白修飾類型,共M列,使用最大最小值歸一化使數據在[0,1]區間,每列數據表示組蛋白修飾與染色質產生作用的可能性。
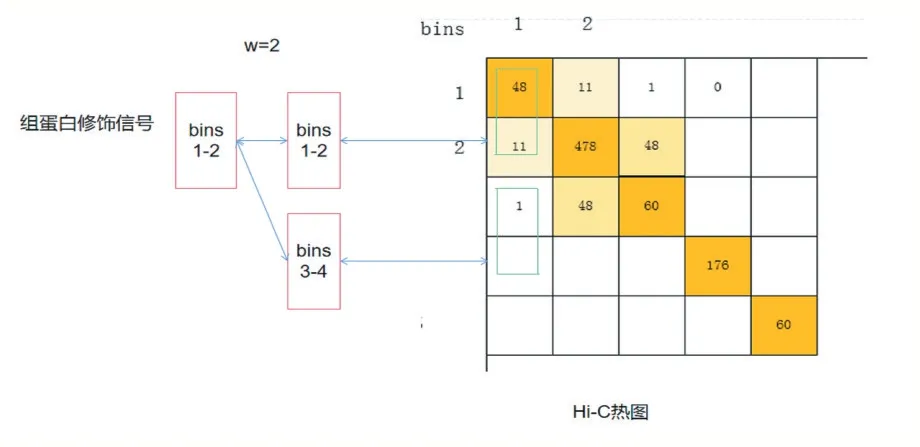
圖1 Hi-C交互作用熱圖
2.2 Hi-C預測模型
神經網絡介紹:卷積神經網絡(CNN)一般用于圖像處理或自然語言處理等高維特征自動提取,可學習到平移不變性等特征,在生物信息研究上也廣泛應用。利用CNN 可以快速提取相應染色質交互的相關因子特征序列,構建交互作用概率分布輸入。在全連接層部分,使用循環神經網絡考慮染色質上下游數據帶來的影響,并使所有神經元之間的參數共享,這些參數針對染色質基相互作用進行了優化,可生成用于不同組蛋白修飾的最佳濾波器,均方誤差(MSE)作為損失函數,平均絕對誤差(MAE)作為目標函數。對輸入輸出數據采用取對數值獲取[-0,1]區間值域范圍,并作為神經網絡模型的標簽和樣本,可理解為對每段染色質交互作用的可能性預測。
本文提出的模型基于每對序列對應的方式,對矩陣中數據一一預測,如圖3 所示,使用一個w 大小的窗口來獲取每個交互基因距離下的所有數據,由于Hi-C矩陣中的對稱特性,交互數據可以按列或行獲取。因為染色質的交互和高維結構受到堿基段的上下游影響,有明顯的區域性,對于輸入的組蛋白修飾序列數據,每個交互位點的上下游各一個堿基段作為輸入,共三個堿基段長度。因此,每個樣本關注w 大小的染色質交互作用,使用x-1 到x+1 段(x 為交互作用發生的位置)的3 個長度的組蛋白修飾作為輸入來預測相應的Hi-C 交互作用情況。本文對模型輸入部分進行劃分,構建一個輸入模塊獲取兩對不同位置的輸入數據,此模型將在Hi-C 矩陣中的對角線區域附近生成每個bin 的交互作用數據。最后結果用熱圖重建方法來還原預測矩陣。窗口大小w 的選取,可使用的Hi-C 矩陣為10kb(104)分辨率下的實驗數據,設定w=50,因此基因組距離為500kb,即每個堿基段包含500k(500×103)的堿基。這樣可以觀察交互作用密集區域,排除交互作用發生不明顯的區域和較遠距離的稀疏數據,使得預測結果更有價值。
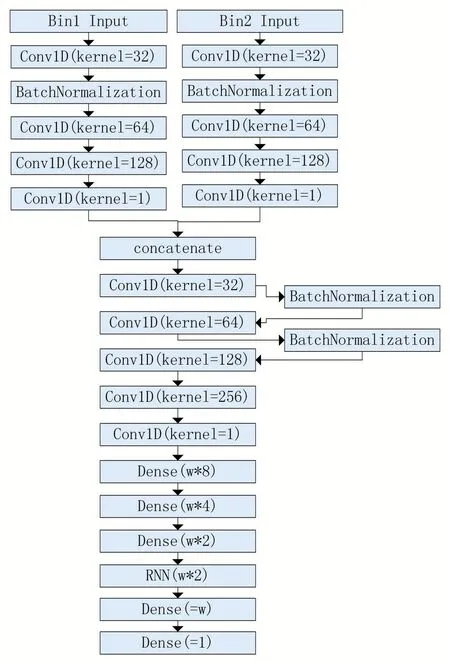
圖2 染色質交互作用預測神經網絡模型結構圖
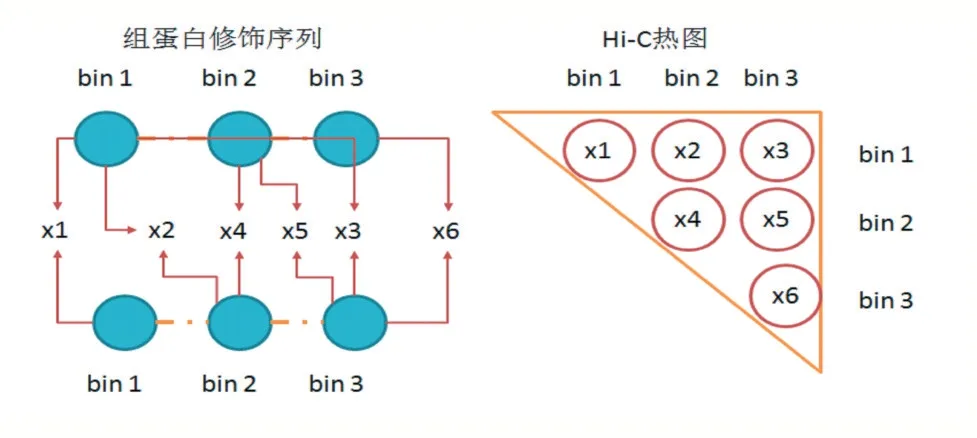
圖3 兩段bins輸入
模型預測過程包括三個階段,第一階段為輸入數據的卷積和特征聚合階段,對每兩對輸入的組蛋白修飾信號矩陣,通過多層一維卷積神經網絡獲取其多種類型修飾的一維聚合特征序列,代表不同修飾類型共同作用的綜合權重分布。第二階段為拼接層,將兩對輸入構成二維矩陣,再次利用卷積神經網絡進行不同位置間的交互作用影響特征提取。最后一層為全連接層和循環神經網絡層,利用堿基上下游序列的影響,轉化為時序序列的前后特征,來預測相應染色質堿基段的交互作用結果。由于神經網絡預測過程中是針對各個位點進行的,需要根據位置和對稱性重建矩陣結果進行對比評估,預測結果中可得到多段w 大小的預測數值排列,根據提取過程的順序可以依次對應到交互作用發生的位置上,并使用重構算法將其恢復成數值矩陣。因此,最終結果也是對角線區域在指示基因組距離上的完整矩陣,并存在一定的拓撲結構區域。
重構矩陣熱圖偽代碼:
M 為最后結果矩陣
For i in N:
if i <N-w+1:M[i,i:i+w]=Input[1:w];M[i:i+w,i]=M[i,i:i+w]
else:x=N-I;M[i,i:i+w]=Input[1:x];M[i:i+w,i]=M[i,i:i+w]
3 結果
我們對模型進行了多次訓練,設定超參數為訓練輪次設定為30 輪,批次大小每次100 個樣本,優化器是RMSProp。訓練完畢后可以獲得一維濾波器的各項權重,表示每對固定組蛋白修飾序列的綜合作用特征。最終結果分布表示序列相關性和矩陣相似性。
最終結果分別使用線性相關和圖像相似性進行分析。數據集使用GEO 數據庫中的GSE63525 訪問代碼獲取的10kb 分辨率的IMR90 原始交互作用數據,重構成交互作用矩陣。其中訓練集使用1-17 號染色體的Hi-C 樣本,測試集使用18-22 號染色體的Hi-C 樣本。最后結果顯示,在500kb 的基因組距離下,對Hi-C 樣本進行預測結果分析。線性相關性分析中測試集PCC 最好達到0.85,SPCC 最好達到0.8,SSIM 的測試樣本中最好可達到0.98。
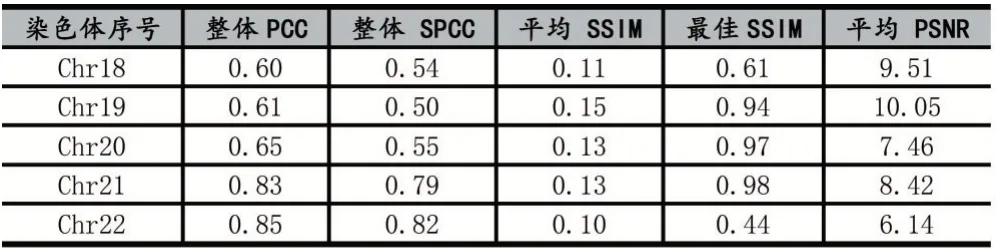
表1 細胞類型IMR90 染色質編號的測試數據集為染色體18 號-22 號/基因組距離(500kb)
4 結語
本文對染色質交互作用的二維結構與表觀遺傳學數據組蛋白修飾信號進行了相關性預測研究,提出了一種基于組蛋白修飾信號序列數據交叉預測染色質交互作用的方法。結果表明,組蛋白修飾信號在染色質相互作用中可起到重要作用,也為染色質結構預測在深度學習和不同數據上提供了一種可行路線。
對于人類細胞類型數據,具有數據量大,交互作用矩陣數據稀疏,結構作用域難以預測等特點,本文提出的方法針對稀疏數據進行了基因距離篩選,一定程度解決了數據稀疏性和不相關數據干擾的問題,并對每對數據進行分別預測,減少了不同樣本差異帶來的訓練過擬合影響。高維染色質結構與染色質組成的內部物質高度相關,可以通過其他組成數據進行推測。

圖4 chr22 16mb-18mb 上的交互
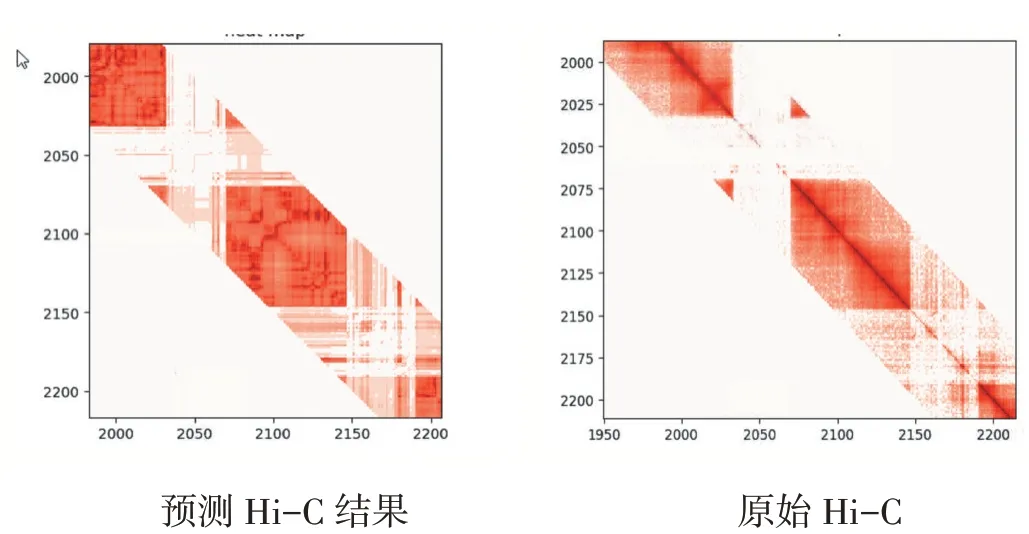
圖5 chr22 20mb-22mb 上的交互
本文的深度學習模型,并應用了不同的數據集和復雜的神經網絡層結構,從組蛋白修飾推測染色質數據。結果表明了模型的能力以及組蛋白修飾的方向,如何影響染色質組織。但方法使用數據類型不豐富,染色質結構不止與組蛋白修飾信號相關,輸入樣本數據特征仍然可以增加,模型預測數據在高維結構的留存性上仍有待提高的。
猜你喜歡
小獼猴智力畫刊(2023年4期)2023-04-23 08:49:58
哲學評論(2021年2期)2021-08-22 01:53:34
中華詩詞(2019年7期)2019-11-25 01:43:04
模具制造(2019年3期)2019-06-06 02:10:54
中學生數理化·高一版(2018年1期)2018-02-10 05:20:03
影視與戲劇評論(2016年0期)2016-11-23 05:26:01
七彩語文·寫字與書法(2016年7期)2016-07-28 21:40:22
七彩語文·寫字與書法(2016年6期)2016-07-15 19:36:34
人間(2015年21期)2015-03-11 15:23:21
現代企業(2015年9期)2015-02-28 18:56:50
