基于氣相色譜-串聯質譜技術測定植物組織中糖與糖醇
2021-05-10 04:45:06堅乃丹李文麗張祝莉朱霞鮮學海牛伊寧
食品與發酵工業 2021年8期
堅乃丹,李文麗,張祝莉,朱霞,鮮學海,牛伊寧*
1(甘肅省干旱生境作物學重點實驗室,甘肅 蘭州,730070)2(臨夏市農產品質量安全中心,甘肅 臨夏,731100)
糖類化合物主要是多羥基的醛或酮及其縮聚物和某些衍生物,源自植物光合作用的初級產物對生物組織結構及生命活動起著重要作用,廣泛存在于自然界[1]。植物細胞中糖和糖醇作為供能代謝基質,具有調控生長、發育、繁殖和免疫等生理功能,參與植物體內三羧酸循環、糖酵解等基礎代謝過程;同時作為信號分子調節相關基因的表達和酶的活性、清除多余自由基和調節植物細胞滲透壓從而提高植物抗逆性[2]。因此植物與糖類互作機制及植物體內糖結構與生物學功能的相關性及其作用機理已成為新研究熱點之一[3],但是植物體中低分子質量的糖與糖醇的濃度較低,所以亟需建立一種準確高效的糖與糖醇定量分析方法。
目前糖類化合物分析方法有高效毛細管電泳法、陰離子交換色譜法、高效液相色譜法、凝膠滲透色譜法、氣相色譜法、質譜法及核磁共振法等[4-9]。其中氣相色譜法樣品用量少,分離能力強,適用于檢測沒有使用限量規定的糖化合物,但其只能通過保留時間進行定性和定量分析,檢測結果的準確性易受復雜基質干擾[10]。質譜法具備高分辨率和高靈敏度等特點,可獲得化合物的結構信息,并且推算化合物的分子質量,對于鑒別有機物分子具有重要作用[11]。氣相色譜-串聯質譜法(gas chromatography-tandem mass spectrometry,GC-MS/MS)兼具色譜的高分離效能與質譜高準確性的特點,特別多級質譜串聯(MS/MS)技術是通過對分子離子和其被碎裂后獲得的碎片離子2次掃描監測以進行數據分析,可提供清晰的分子離子與碎片離子之間的關系,增加定量準確性,廣泛應用于復雜基質中痕量物質的檢測[12]。丁潔等[13]利用氣相色譜法測定金銀花中含有的12種糖和糖醇并繪制了其指紋圖譜。BOLDIZAR等[14]采用氣相色譜-質譜聯用法對香芹果實及葉片組織中的糖和糖醇進行定量分析。但是還尚未見文獻報道使用氣相色譜-串聯質譜法同時測定多種糖與糖醇的方法。
1 材料與方法
1.1 材料與試劑
原料:黑果枸杞購自青海格爾木市大格勒鄉;蘆薈采自室內栽培植株。
試劑:吡啶、乙酸酐、鹽酸羥胺均為分析純,成都科隆化學品有限公司;色譜級氯仿,青島天澤生物技術有限公司;側金盞花醇(Ado)、葡萄糖(Glu)、半乳糖(Gal)、肌醇(Ino)、甘露醇(Man)、山梨醇(Sor)、蔗糖(Suc)標準品(純度均大于98%),成都樂美天醫藥科技有限公司。
1.2 儀器與設備
GC-MS/MS三重四極桿氣相色譜質譜聯用儀7890B-7000D(配有EI離子源和NIST MS數據處理系統),美國Agilent公司;DF-110S集熱式磁力攪拌器,常州瑞華制造有限公司;毛細管柱DB-1701、DB-23、HP-5MS(均為30 m × 0.25 mm,0.25 μm),美國Agilent公司;冷凍臺式真空離心濃縮儀,日本京東理化公司。
1.3 標準溶液配制
精確稱取側金盞花醇2 mg,用無水吡啶溶解并定容至10 mL作為內標。另精確稱取其他糖和糖醇標準品(葡萄糖、半乳糖、肌醇、甘露醇、山梨醇、蔗糖)2 mg,用無水吡啶溶解并定容至10 mL。分別吸取各標準品1.4、1.2、1.0、0.8、0.6、0.4、0.2、0.1、0.05、0.025 mL于離心管中,分別加入內標溶液0.5 mL,再以無水吡啶補充至2 mL。于4 ℃冰箱避光保存。
1.4 樣品提取
1.4.1 氯仿-甲醇水溶劑提取法[15]
稱取0.2 g植物樣品進行液氮研磨,加入4 mL提取液(甲醇-氯仿-水體積比12∶5∶3),充分搖勻,再加入4 mL去離子水,混和均勻,離心取上清液至試管,旋轉蒸干,補充2 mL吡啶,于4 ℃冰箱保存。
1.4.2 超聲輔助提取法
稱取0.2 g植物樣品進行液氮研磨,置于20 mL的離心管,加入5 mL去離子水,10 mL的無水乙醇,置超聲儀超聲30 min,離心,4 000 r/min,去上清。殘留不溶物用10 mL 80%的乙醇水溶液洗滌、離心,去上清,加入10 mL水,超聲提取30 min,重復2次。冷卻至室溫,離心取上清液,旋轉蒸干,補充2 mL吡啶,于4 ℃冰箱保存。
1.5 標準品及樣品乙酰化處理[16]
向待測樣品及標準品溶液加入20 mg鹽酸羥胺充分溶解,置于90 ℃水浴鍋反應30 min,冷卻至室溫,加入1 mL無水乙酸酐,再次于90 ℃下反應30 min,冷卻后加入1 mL去離子水攪拌,然后用氯仿溶液萃取3次,合并氯仿層,旋轉蒸干,加入1 mL氯仿溶解,進行分析檢測。
1.6 分析條件
1.6.1 色譜條件
色譜柱:HP-5MS色譜柱(30 m×0.25 mm,0.25 μm);進樣口溫度:300 ℃,載氣:高純氦氣;碰撞氣:高純氮氣,純度≥99.999%;柱流量:1 mL/min;進樣方式:分流進樣200∶1;輔助加熱溫度280 ℃;升溫程序:初始溫度120 ℃保持3 min,以20 ℃/min的速率升至315 ℃,保持5 min,進樣量1 μL。
1.6.2 質譜條件
離子源:EI源;離子源溫度設置230 ℃,四極桿溫度設置150 ℃;電離能量設置70 eV,溶劑延遲8 min;掃描類型:多重反應監測(multiple reaction monitoring,MRM)模式。7種乙酰化糖和糖醇的保留時間、前級離子、子離子和碰撞能量等參數見表1。
2 結果與分析
2.1 質譜條件
MRM模式是利用使用第一級四極桿(MS1)來選擇某一質量的前級離子,將其輸送至六極桿碰撞反應池進行碎裂,然后通過第二級四極桿(MS2)來監測特征離子碎片。由于MRM模式在第二級四極桿階段可去除許多化學背景,出現與碎裂離子質量完全相同的同種干擾物機會較少,所以相比單四極桿,三重四極桿質譜定量復雜基質中的低濃度目標物會更加減少化學噪音干擾[17]。在離子源EI模式下對7種揮發性乙酰化糖和糖醇首先進行全掃描一級質譜分析,掃描范圍35~660 amu,在所得目標物質譜圖中選擇質荷比較大且響應強度較高的離子碎片作為前級離子。過低的碰撞能量會導致目標物質無法被電離,而過高的碰撞能量會導致目標物質被轟碎,所以將碰撞能量范圍設置5~50 eV(每5eV一個間隔)確定最優碰撞能量,對選定的前級離子峰進行產物離子掃描。前級離子進入二級質譜,將發生斷裂或重排等反應產生不同的離子碎片,選取其中響應強度最高的離子作為定量離子,響應強度次之的為定性離子。最后采用MRM模式對目標化合物進行定性定量分析。優化后的質譜分析參數見表1,目標化合物MRM色譜圖見圖1。
2.2 色譜條件
進樣口汽化溫度遠低于化合物沸點時,化合物連同少量的樣品基質會殘留在襯管中,可導致后續進樣被污染。選取進樣口溫度分別為260、280、300、320 ℃進行樣品檢測。對濃度為80 mg/L的混標進行分析,實驗結果表明,7七種乙酰化糖和糖的峰面積最大值對應的進樣口溫度有所不同,但其峰面積均隨進樣口溫度的升高逐漸上升并在300 ℃時基本恒定,故選擇的進樣口溫度為300 ℃。
實驗分別考察了弱極性的HP-5MS、中極性DB-1701和強極性DB-23三種毛細管柱對7種糖類化合物的分離效果。結果表明,乙酰化蔗糖在柱溫315 ℃可順利分離,而DB-23與DB-1701的毛細管柱極限溫度分別僅達到250 ℃與280 ℃,只可分離其他6種乙酰化糖。HP-5MS毛細管柱極限溫度是325 ℃,同時具有超高惰性且柱流失低,可獲得較好的峰形和較高的信噪比,因此選取色譜柱HP-5MS為本次實驗的分析柱。
乙酰化甘露醇和山梨醇是同分異構體,在SCAN模式下程序升溫分離度較差,采用MRM模式分離效果顯著改善,與周洪斌等[18]利用降低柱箱升溫速率使得乙酰糖分離方法相比,可充分節約檢測時間,提高效率。經過調試初始柱溫、升溫速率、分流模式等參數,7種目標物在15 min內達到基線分離,且檢測響應相對較高。圖2為MRM模式下7種目標化合物的總離子流色譜圖。

1-側金盞花醇;2-葡萄糖;3-半乳糖;4-肌醇;5-甘露醇; 6-山梨醇;7-蔗糖圖2 七種乙酰化糖和糖醇的總離子色譜圖Fig.2 Total ion chromatogram of the 7 aceylated sugars and sugar alcohols
2.3 樣品提取方法選擇
本次實驗采用氯仿-甲醇水溶劑提取法與超聲波輔助提取法分別提取枸杞樣品中的糖與糖醇,結果如圖3所示。采用氯仿-甲醇水溶劑提取法相較超聲波輔助提取法所得葡萄糖、肌醇與蔗糖含量略高,但是半乳糖、甘露醇與山梨醇含量略低。2種不同的提取方式均可以提取出糖或糖醇物質且提取效果差異相對較小,考慮到氯仿-甲醇水溶劑提取法操作比較方便、提取液顏色淺,提取用時短,今后實驗可選擇氯仿-甲醇水溶劑提取法制備樣品溶液。

圖3 不同提取方法對6種糖和糖醇提取量的影響Fig.3 Effect of different extraction methods on the content of six sugars and sugar alcohols
2.4 MRM方法驗證
目標分析物的色譜峰峰型對稱且沒有基質干擾存在時,可根據保留時間對被分析物進行定性,根據定量離子對的峰面積定量。將1.3項下的標準系列工作液按照優化的分析方法進樣測試,為避免前處理過程中出現操作誤差,以其他標準品與內標側金盞花醇的峰面積比為縱坐標,以標準品濃度(mg/L)為橫坐標繪制標準曲線,并計算回歸方程和相關系數。對標準溶液進行逐級稀釋,分別以信噪比(S/N)為10和3時的溶液濃度作為定量限和檢出限。結果表明,各化合物線性關系良好,相關系數(R2)均大于0.997,檢出限在0.013~0.12 mg/L,定量限在0.045~0.37 mg/L。本方法的線性范圍廣,靈敏度高。
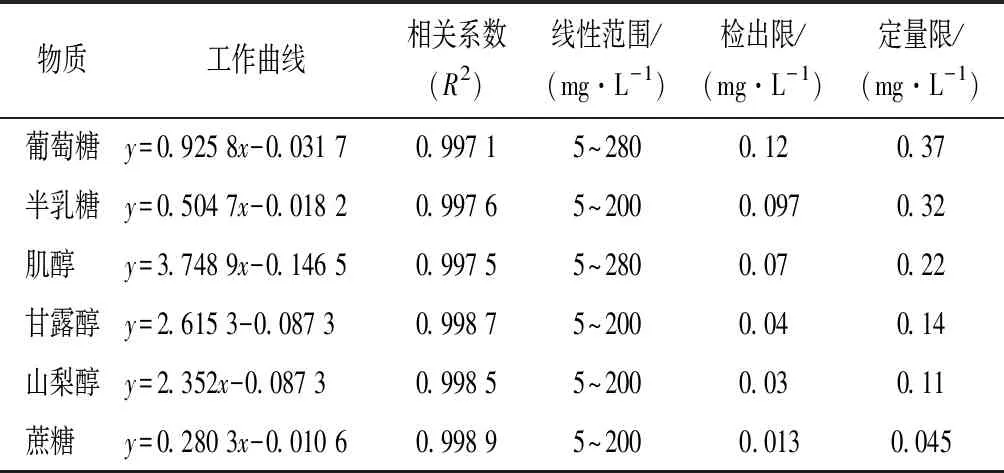
表2 乙酰化糖和糖醇的工作曲線、相關系數、檢出限和定量限Table 2 Regression equations, correlation coefficients,limits of detection and limits of quantification of 6 acetylated sugars and sugar alcohols
選取1組枸杞樣品,每份0.2 g,在前處理時分別添加3種不同水平含量的糖和糖醇標準品進行加標回收實驗,重復測定6次,如表3顯示,每份處理回收率為95.38%~102.6%,相對標準偏差(RSD)為1.28%~3.42%(見表3),該方法精密度良好,達到實際樣本檢測要求。
2.5 實際樣品測量
天然產品藥用等級的一個指標是具有藥理活性的多糖含量[19]。黑果枸杞屬于茄科枸杞屬旱生灌木植物,其果實味甘性平,含有大量藥理活性的糖類、類胡蘿卜素、多酚類等物質,藥食兩用[20]。蘆薈是百合科蘆薈屬多年生常綠草本植物,具有降血糖,抗腫瘤,抗艾滋以及增強免疫等多種功能,其營養功效主要來自大量的多糖成分。蘆薈不同部位含糖量不同,根莖部位的多糖含量較多,葉皮次之,然后是全葉[21]。故本次實驗選取枸杞果實與蘆薈莖葉部位作為樣本,采用氯仿-甲醇水溶劑提取法處理樣品,按照已建立的方法測定樣品中糖和糖醇的含量,如表4所示。結果表明,黑果枸杞中葡萄糖含量最高,蔗糖含量最低。蘆薈中則是半乳糖含量較高,山梨醇含量最低。采用GC-MS/MS多重反應監測模式可以準確地測定植物中糖化合物含量,得到清晰準確的色譜峰。

表3 MRM模式下方法的回收率和精密度Table 3 Recoveries and precisions at MRM mode

表4 植物樣本中糖和糖醇含量
3 結論
糖類的乙酰化衍生主要經歷肟化和乙酰化兩步反應。其中肟化反應條件要求較高,反應體系內各組分之間互有協同衍生化反應的影響,需在適宜的溫度和反應持續時間下才能順利向正向反應方向進行[22]。目前對糖類化合物進行乙酰化方法通常使用鹽酸羥胺和乙酸酐,1-甲基咪唑或吡啶作為催化劑和溶劑。本次實驗中對于樣品進行肟化處理時采用吡啶作為溶劑,萃取過程正常,但是采用1-甲基咪唑作為肟化反應溶劑,在萃取中糖溶液不發生分層現象且溶液顏色呈現為黑色,與文獻報道[23]存在差異,可能是反應處理時間及其使用量存在不當問題,需要進一步探究。
糖和糖醇乙酰化生成具有揮發性且沒有異構峰的糖類衍生物進行氣相色譜分離,從而排除了糖類同分異構體的干擾。HP-5毛細管柱在反復使用3個月后,乙酰化糖和糖醇的保留時間位移均在±0.05 min內,考慮儀器誤差,認為該方法重現性變化范圍符合技術要求。7種乙酰化糖和糖醇通過毛細管色譜柱分離后進入質譜檢測,在MRM模式下被分配到不同的通道中,保留時間相對較近的目標物也能完成較好的分離效果。通常多種化合物若在色譜中取得良好的分離效果用時較長,本次實驗采用的串聯質譜法中多反應監測模式有效地改善了這一不足之處[24]。
本實驗通過乙酰衍生化的方法將側金盞花醇、葡萄糖、半乳糖、肌醇、甘露醇、山梨醇和蔗糖轉變為可揮發性物質,結合內標法,建立氣相色譜-串聯質譜法同時對多種衍生糖化合物進行定性定量分析。實驗采用的多重反應監測模式既克服基質效應對響應信號的干擾,又提高實驗準確度。該方法樣品檢測用量少,結果穩定且靈敏度高,可應用于檢測植物組織中微量的糖化合物,同時也為檢測其他樣本中的糖化合物提供更多的選擇。
