核磁共振波譜法結合氧彈燃燒測定茶葉中的總氟
2021-05-10 06:47:56朱正偉朱松松王會霞
食品與機械 2021年4期
周 密 韓 智 朱正偉 朱松松 王會霞 江 豐 朱 芊
(1. 湖北省食品質量安全監督檢驗研究院,湖北 武漢 430075;2. 湖北省食品質量安全檢測工程技術研究中心,湖北 武漢 430075)
氟是人體重要的微量元素之一,參與人體的許多生命活動[1]。氟適量攝入有益于人體骨骼和牙齒的鈣化,增加骨骼的強度并維持口腔健康[2-3],但攝入過量則會導致全身慢性蓄積性中毒,形成“地氟病”,輕者形成氟斑牙,重者造成氟骨病,嚴重影響生長發育,危害身體健康[4-5]。茶樹[Camelliasinensis(L.) O. Kuntze]有較強富集氟的能力[6-8],葉片是茶樹富集氟的主要器官,盡管不同質量、不同產地、不同品牌的茶含氟量不同[9],但隨著茶葉葉齡的增加,葉片中氟含量也逐漸升高[10-14],老葉含氟量更是高出嫩葉近10倍[15]。隨著近年來消費提檔升級,有學者[16]將超微茶粉直接開發成固體飲料,提高茶葉中有效成分的利用率,以滿足市場多元化需求,但缺少對該類產品中總氟含量的關注,如若生產商使用老葉或劣質茶葉為原料加工生產,必定增加食用人群對于氟的攝入量,對身體健康構成威脅。
茶葉中氟的測定前處理方法主要有浸泡法[17]、灰化堿熔法[18]和氧彈燃燒法[19-20]。現行國標[17]中茶葉氟的測定方法主要是針對水溶性氟,但植物細胞內的氟難以被完全提取,無法準確反映產品中總氟含量;灰化堿熔法在高溫、開放的體系中進行,處理時間長達3~5 h,氟易損失[20];氧彈燃燒使茶葉(粉)樣品在密閉富氧的條件下反應,將氟元素全部轉化為無機態氟[20],樣品損失小,適合茶葉總氟含量測定[19]。
定量核磁共振技術(quantitative nuclear magnetic resonance,qNMR)已被廣泛用于天然產物表征[21-22]、參考物質質量控制[23-24]和藥物分析[25-26]中,其中,核磁共振氟譜(19F quantitative nuclear magnetic resonance,19F qNMR)具備化學位移范圍大、靈敏度高、結構近似的化合物不易出現峰重疊[27]等優勢,在定量分析中應用廣泛。研究擬選擇19F qNMR技術,以三氟乙酸為內標,測定茶葉中氟含量,可為茶葉及其制品中總氟的數據監控提供技術支撐。
1 材料與方法
1.1 材料與儀器
1.1.1 主要儀器設備
全數字化超導核磁共振譜儀:AVANCE Ⅲ 600 MHz型,德國Bruker公司;
超純水系統:Purelab Chorus型,英國ELGA公司;
刀式搗磨儀:GM 300型,德國Retsch GmbH公司;
強制對流通用烘箱:UF 110 PLUS型,德國Memmert公司;
電子天平:ME 204型,梅特勒—托利多儀器(上海)有限公司;
粉末壓片機:FW-5型,天津市拓普儀器有限公司;
鹵素分析前裝置:WBLS-1型,含充氧裝置、點火裝置,萬邦儀表科技有限公司;
恒溫振蕩水浴鍋:WNB 29型,德國Memmert公司;
超聲波雙頻清洗機:SB25-12DTS型,寧波新芝生物科技有限公司。
1.1.2 材料與試劑
重水:純度99.9%,Cambridge Isotope Laboratories, Inc;
高純氧:純度99.999%,四川天一科技股份有限公司武漢供氣分公司;
氟化鈉、碳酸銨、檸檬酸鈉、乙二胺四乙酸二鈉(EDTA-2Na)、三氟乙酸:分析純,上海阿拉丁生化科技股份有限公司;
測試茶葉樣品:湖北省食品質量安全監督檢驗研究院;
茶葉(綠茶粉)中氟質控樣品(P24307):廣州譜恩科學儀器有限公司。
1.2 試驗方法
1.2.1 譜儀測定條件 在前人[28]的基礎上,根據儀器硬件條件,對譜儀測定條件適當修改。脈沖序列為zgfhigqn,采樣點數64 000,采樣時間0.961 2 s,掃描次數32次,譜寬34 091 Hz,觀察道中心頻率偏置-56 468 Hz,去耦道中心頻率偏置4 801 Hz,脈沖寬度12.0 μs,測定溫度298 K。采用t1irpg脈沖序列,確定譜儀的測定參數延遲時間d1。
1.2.2 內標物選擇及氟標準曲線 稱取0.200 0 g三氟乙酸于100 mL容量瓶中配制成含氟當量為1.0 mg/mL的三氟乙酸溶液,將氟化鈉于120 ℃烘干2 h,準確稱取0.221 0 g,加水溶解,定容至100 mL,搖勻,濃度為1.0 mg/mL,進一步配制質量濃度為2.0,4.0,8.0,12.0,20.0 μg/mL溶液,均含氟當量為10.0 μg/mL 的三氟乙酸溶液。精密移取540 μL標準溶液,60 μL重水于核磁管中,混勻。按1.2.1所述方法采集19F NMR譜圖,考察標物三氟乙酸與目標物氟標準溶液的分離情況,確定內標物;以目標物、內標物峰面積之比為縱坐標,目標物、內標物質量濃度之比為橫坐標,考察線性關系。
1.2.3 樣品前處理 參照文獻[19—20],試樣經搗磨儀粉碎、過40目篩,于80 ℃烘干至恒重,稱取約0.50 g茶葉試樣,壓片后放入氧彈坩堝中,于氧彈反應釜內加入15 mL 20 mmol/L碳酸銨溶液作為吸收液,擰緊氧彈蓋,緩慢充入3.0 MPa純氧,將氧彈置于振蕩水浴中,點火燃燒灰化,充分反應后開啟放氣閥,加入金屬螯合劑,用溫水少量多次潤洗、超聲,定容至50 mL,過0.45 μm濾膜,待測。按1.2.1所述方法采集核磁氟譜,比較加入不同金屬螯合劑(檸檬酸鈉溶液、EDTA-2Na溶液、檸檬酸鈉—EDTA-2Na混合溶液,均含氟當量為10.0 μg/mL的三氟乙酸)對氟譜信號的影響。
1.2.4 數據處理及計算公式 使用Bruker Topspin 3.5軟件對譜圖進行相位和基線校正,對內標及目標峰進行峰面積積分,按式(1)計算積分結果。
(1)
式中:
X——待測物含量,mg/kg;
m——稱樣量,g;
mstd——每50.0 mL含有內標物的質量,μg;
Mx——待測物分子量;
Mstd——內標分子量;
Ax——待測物定量峰信號面積;
Astd——內標物定量峰信號面積;
nx——待測樣品包含的氟原子數,按F-計為1;
nstd——內標物包含的氟原子數,按CF3COOH計為3。
2 結果與分析
2.1 延遲時間d1的選擇
使用NMR定量分析時,一定要保證選擇足夠長的延遲時間d1(≥5×T1),否則對結果影響很大[28]。試驗通過t1irpg脈沖序列,測定三氟乙酸和氟化鈉的延遲時間d1,分別為3.0 s和1.6 s,故使用三氟乙酸為內標測定樣品中氟離子時,選擇d1為15.0 s。
2.2 內標物及定量峰的選擇
使用內標進行定量分析過程中,需要保證內標物質峰與待測物質峰良好分離。加入一定量的內標溶液及氟化鈉標準溶液,加入10% D2O,于1.2.1所述條件進行19F NMR測試,測試譜圖見圖1。
圖1中δ=-120的尖銳單峰為氟化鈉信號峰,δ=-75的尖銳單峰為三氟乙酸信號峰,與文獻[28]相符,被測物質的定量峰與內標物的定量峰完全分離,說明使用三氟乙酸作為內標具有可行性。
2.3 線性關系的確定
取質量濃度為2.0,4.0,8.0,12.0,20.0 μg/mL溶液(均含氟當量為10.0 μg/mL的三氟乙酸溶液)進行線性關系考察。以樣品積分面積與內標物的積分面積比(S1/S2)為縱坐標,以樣品的濃度與內標物的質量濃度比(C1/C2)為橫坐標,結果見圖2。
由圖2可知,當樣品質量濃度為2.0~20.0 μg/mL時,S1/S2與C1/C2呈良好的線性關系,回歸方程為y=1.027 1x+0.037 8,回歸系數R2=0.999 5,進一步說明使用三氟乙酸作為內標對樣品中氟進行定量分析是可行的。
2.4 金屬螯合劑的選擇
氟離子作為性質活潑的陰離子之一,易與Ca2+、Fe3+、Mg2+、Al3+等金屬陽離子形成絡合物從而影響測定,GB 19965—2005通過添加檸檬酸鈉屏蔽金屬離子,也有學者[29]通過添加EDTA-2Na絡合金屬Al元素。試驗選取2.50%檸檬酸鈉溶液和1.25% EDTA-2Na溶液作為金屬螯合劑,考察金屬螯合劑添加與否、不同金屬螯合劑添加量對測定結果的影響。
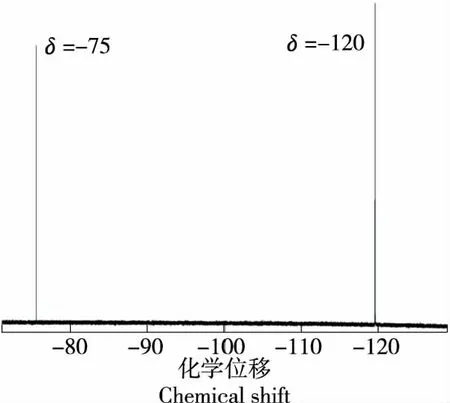
圖1 內標物與目標物的19F qNMR譜圖
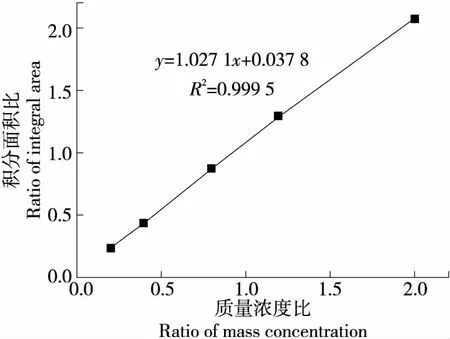
圖2 積分面積與質量濃度線性關系
結果表明,在不添加金屬螯合劑和只添加1.25% EDTA-2Na溶液5 mL時,經過氧彈燃燒后的樣品在δ=-119處無目標峰出現,如圖3(c)和圖3(d)所示;圖3(b)中,在添加2.50%檸檬酸鈉溶液5 mL時,樣品在δ=-119處出現目標峰,但峰型較差,定量困難;而在添加金屬混合螯合劑(1.25% EDTA-2Na溶液、2.50%檸檬酸鈉溶液)5 mL時,樣品在δ=-119處目標峰峰型良好,如圖3(a) 所示,通過增加掃描時間,即可對目標物質進行定量分析。這可能是由于不同螯合劑對不同價態金屬螯合作用不同,進一步將性質活潑的絡合態氟元素,從與金屬絡合的狀態下釋放出來。另外,增加金屬螯合劑至10 mL 時結論一致,但不同金屬混合螯合劑添加量對峰響應強度有一定影響。
進一步添加0.0,2.5,5.0,7.5,10.0 mL不同水平的金屬混合螯合劑,比較目標峰響應響度,見圖4。結果表明,未添加金屬混合螯合劑時無目標峰響應,金屬混合螯合劑添加至5.0 mL后響應強度趨于穩定,繼續添加7.5,10.0 mL 金屬混合螯合劑,信號響應強度無明顯變化,說明在試樣稱樣量為0.5 g水平下,金屬混合螯合劑與溶液中的金屬離子充分絡合。故氧彈反應完全,添加5.0 mL金屬混合螯合溶液為宜。
2.5 方法學考察
2.5.1 最低檢測濃度 依據《中國藥典》(2015版)第4部通則,以S/N≥3時確定最低檢測濃度,在1.2.1所述條件下,當δ為-70~-130,測定時間為8 min 31 s時,最低檢測濃度為2.0 μg/mL。
2.5.2 重復性試驗 選取茶葉(綠茶粉)中氟質控樣品(P24307)、磚茶樣品,按1.2.3所述方法,重復測定6次,氟質控樣品典型測試譜圖如圖5所示,定義三氟乙酸峰面積為1.000 0(原始積分數據3 568 081.9),目標峰氟離子面積為0.293 7(原始積分數據1 047 966.5)。測試結果如表1所示,其相對偏差為6.04%~6.98%,方法重復性良好。另外,茶葉(綠茶粉)中氟質控樣品(P24307)中位值為319 mg/kg,|Z|≤2.0時測試值范圍為258~380 mg/kg,測試值為301 mg/kg,測試結果滿意。

圖3 螯合劑對響應強度的影響
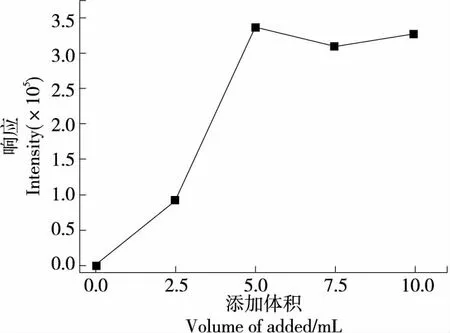
圖4 不同體積螯合劑對響應響度的影響
2.5.3 精密度試驗 隨機選取茶葉樣品,通過19F qNMR平行測定總氟含量6次,定義三氟乙酸峰面積為1.000 0,對目標峰積分,計算相對峰面積,6次積分面積結果相對偏差為2.50%,表明使用19F qNMR測定總氟含量具有較高的精密度。
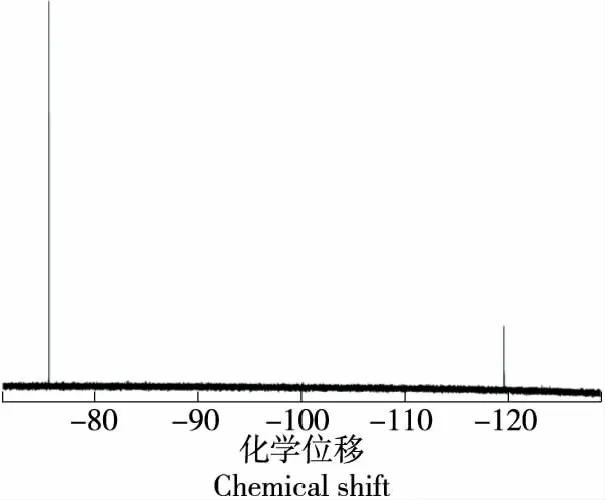
圖5 典型樣品測試譜圖
2.5.4 回收率試驗 茶葉樣品基質(1 226 mg/kg)中添加氟含量分別為200,400,2 000 mg/kg三水平的標準溶液,每個水平平行測定3次,計算回收率和相對偏差,結果如表2所示,加標回收率為96.3%~107.0%,相對偏差為4.20%~6.07%。
3 結論
采用氧彈燃燒對樣品進行前處理,以三氟乙酸為內標,在無外部校準曲線的情況下,結合19F qNMR對茶葉中的總氟進行快速定量分析,填補中國使用19F qNMR測定茶葉樣品總氟含量的技術空白,30 min內可完成單個樣品分析,檢測效率高,方法準確、穩定性良好,具有一定的實用性。在對茶葉樣品總氟含量的分析測定中,可以考慮添加混合螯合劑,進一步釋放與金屬離子絡合的氟元素。該方法在靈敏度上存在一定的局限,后續研究中可考慮優化譜儀脈沖條件、采用變溫試驗等方式來提高檢測靈敏度。

表1 重復性試驗

表2 回收率試驗
