液相色譜-原子熒光光譜聯用法測定土壤砷鉻銻硒元素價態
2021-05-08 03:09:48薛佳
巖礦測試 2021年2期
關鍵詞:實驗
薛佳
(福建省地質測試研究中心,福建 福州350003)
重金屬的環境效應評價是生態地球化學調查的一項重要內容,早期污染物總量被認為是影響環境的主要因素,20世紀末人們認識到化學元素的生物毒性或益處、在生物體的代謝機制都與其在介質中的形態分布有著密切的關系[1]。土壤中重金屬主要以無機態形式存在,有機態含量較少,因此對無機價態分布的研究更能反映污染狀況。作為重要的生態評價指標,砷、鉻、銻和硒的價態研究成果日益增多,但是價態不穩定性和價態之間易轉化,導致測試難度大,國家標準或地質行業標準尚未出臺。地質行業標準《生態地球化學評價樣品分析技術要求》(DD2005-03)早已認識到土壤元素價態測定的重要性,在該標準附錄B中給出了價態測定的指導建議,但未制定具體操作規程,實際檢測和評價工作尚無統一標準。目前國土資源大調查中的多目標生態調查評價仍采用元素全量進行評價,不易對實際污染狀況作出準確判斷。建立適合生態地球化學調查評價的土壤樣品元素價態分析方法,對提高生態地球化學、環境地球化學、特色高效農業和地質安全評價工作水平具有十分重要的意義。
價態測定方法主要分為非色譜法和色譜法。非色譜法出現較早,一般采用差減法計算結果,如依靠控制條件測定甲價態含量和元素總量,差減得到乙價態含量。價態分離主要采用直接測定法[2]、控制酸度法[3]、加入掩蔽劑屏蔽法[4-6]、選擇性還原法[7-8],或采用吸附、萃取或共沉淀等富集分離的方法,其中吸附法應用較多,吸附劑有離子交換樹脂[9-11]、巰基棉[3]、硅膠[12]和磁性納米四氧化三鐵[13]等。由于早期大型儀器聯用技術尚未普及,DD2005-03中推薦采用非色譜差減法,主要問題是操作繁瑣,測定次數多工作量大,在測定甲價態時乙價態不能完全被掩蔽,可能造成甲價態測定值偏高,而且無法排除有機形態的干擾,導致結果出現偏差。近二十年來色譜-光譜聯用技術發展迅速,如液相色譜、離子色譜、毛細管電泳等高效分離儀器和電感耦合等離子體質譜(ICP-MS)、原子吸收光譜(AAS)、原子熒光光譜(AFS)等高分辨率檢測儀器的聯用[14-36],排除復雜基體的干擾,一次進樣可同時測定多個組分,過程簡便,成為元素形態和價態分析的理想方法。色譜差減法的應用[22-23]也有報道。色譜法可實現兩價態同時測定,結果更準確,但該方法的主要問題是儀器裝置復雜,成本較高。而一些國產儀器如液相原子熒光形態分析儀(LC-AFS)的性價比較高,適用于日常檢測[37-38]。
本研究旨在建立一套經濟高效的土壤水溶態和離子交換態提取液中As、Cr、Sb、Se價態測定的方法,以滿足多目標生態地球化學調查評價項目樣品檢測要求。通過大量實驗對比,建立了采用水浴振蕩加熱浸提,液相色譜-原子熒光光譜法(LCAFS)分離和測定As、Sb、Se價態,陽離子交換樹脂分離-電感耦合等離子體質譜法(ICP-MS)測定Cr價態的方法。由于某些離子交換態提取劑的限制,同時還建立了AFS選擇性測定Sb、Se價態的方法,并對離子色譜-ICP-MS聯用法測定As、Cr、Sb、Se價態的方法進行了研究,補充完善了DD2005-03要求的價態測定方法,進而制定適合檢測工作的操作規程,為調查評價工作以及土壤污染修復等工作提供技術支持。
1 實驗部分
1.1 儀器及工作條件
液相原子熒光形態分析儀(北京海光儀器有限公司);離子色譜儀(ICS-90,美國Dionex公司);電感耦合等離子體質譜儀(ICP-MS,美國ThermoFisher公司)。
Millipore超純水機;超聲波清洗器(昆山超聲儀器有限公司);臺式高速離心機(上海安亭精密儀器廠);恒溫水浴振蕩器(天津賽得利斯)。
1.2 標準溶液和主要試劑
As(Ⅲ)單標(1000 mg/L,美國O2si公司,Catalog No.060033-08-01,有效期18月)。
As(Ⅴ)單標(1000mg/L,美國O2si公司,Catalog No.060033-26-01,有效期18月)。
Se(Ⅳ)單標(1000 mg/L,美國O2si公司,Catalog No.060034-08-01,有效期18月)。
Se(Ⅵ)單標(1000 mg/L,美國O2si公司,Catalog No.060034-09-01,有效期18月)。
Cr(Ⅲ)和Cr(Ⅵ)單標(1000mg/L,國家有色金屬及電子材料分析測試中心)。
酒石酸銻鉀(優級純),六羥基銻酸鉀(>99.0%)、磷酸氫二銨、磷酸氫二鉀、磷酸二氫鉀、硼氫化鉀、碘化鉀、鄰苯二甲酸氫鉀、乙二胺四乙酸二鈉、醋酸銨、硝酸銨、氨水、氫氧化鉀、氫氧化鈉、硫脲、抗壞血酸、檸檬酸、甲酸、鹽酸、硝酸、甲醇(色譜純)。所有試劑無特別說明均為分析純。
732陽離子交換樹脂。
超純水(電阻率18.2MΩ·cm)。
1.3 樣品采集
2007年6月于福建省福州市郊菜園果園及野地采集表層土壤樣品共50個,過20目篩(<0.84 mm),經室溫風干混勻后縮分取土壤試樣200g,采用瑪瑙無污染樣品制備機具將樣品粉碎至100目(<0.15mm),裝袋備用。用AFS法分別測定該批土壤中的As、Cr、Sb、Se總量。為觀察浸提價態含量與總量的關系,根據測定結果選取砷和鉻高中低含量有代表性的樣品各5個,編號為:砷N01~N05、鉻N06~N10。銻和硒土壤總量較低,選取含量相對高的樣品,編號為:銻N11~N15,硒N16~N20。
1.4 樣品前處理
準確稱取制備好的土壤樣品2.000g放入50mL離心管中,加20mL去離子水,50℃加熱振蕩30min后,以5000r/min離心10min,倒出上清液,即為水溶態提取液。再往離心管內加入離子交換態提取劑20mL,重復上述步驟制備離子交換態提取液。
As、Cr、Sb和Se元素按DD2005-03的要求使用不同的離子交換態提取劑,分別為:0.6mol/L磷酸二氫鉀溶液、0.3mol/L醋酸銨溶液、0.2mol/L酒石酸溶液和0.1mol/L磷酸二氫鉀-磷酸氫二鉀溶液。
1.5 樣品分離測定方法
1.5.1 液相色譜-原子熒光光譜聯用法測定砷銻硒價態
土壤浸提液經0.45μm水系濾膜過濾,按照以下儀器工作條件用液相色譜-原子熒光形態分析儀進行測定,同時測定空白。
液相色譜工作條件:Dionex IonPac AS19離子色譜柱(250mm×4mm);流速1.0mL/min;進樣量100μL。
原子熒光光譜工作條件:泵速60r/min;載氣流速300mL/min,屏蔽氣流速900 mL/min;爐高8mm。
(1)As(Ⅲ)和As(Ⅴ)的測定
用1.2節中As(Ⅲ)和As(Ⅴ)單標配制2、10、50、100、200μg/L的As(Ⅲ)和As(Ⅴ)混合標準溶液。
流動相:100mmol/L磷酸氫二銨,10%甲酸調節pH 6.0;砷空心陰極燈193.7nm;主電流/輔電流80/40mA;負高壓310V;還原劑:3%硼氫化鉀-0.5%氫氧化鉀;載流5%鹽酸。
(2)水溶態Sb(Ⅲ)和Sb(Ⅴ)的測定
準確稱取0.2743g酒石酸銻鉀溶于水,定容至1000mL,稱取0.2159g六羥基銻酸鉀溶于水,定容至1000mL,配制成Sb(Ⅲ)和Sb(Ⅴ)單標各100mg/L。再用單標配制2、10、50、100、200μg/L的Sb(Ⅲ)和Sb(Ⅴ)混合標準溶液。
流動相:1mmol/L鄰苯二甲酸氫鉀-10mmol/L乙二胺四乙酸二鈉;銻空心陰極燈217.6nm;主電流/輔電流90/45mA;負高壓350V;還原劑:3%硼氫化鉀-0.5%氫氧化鉀;載流7%鹽酸。
(3)Se(Ⅳ)和Se(Ⅵ)的測定
用1.2節中Se(Ⅳ)和Se(Ⅵ)單標配制2、10、50、100、200μg/L的Se(Ⅳ)和Se(Ⅵ)混合標準溶液。
流動相:100mmol/L磷酸氫二銨,10%甲酸調節pH 6.0;紫外消解開啟;還原劑:5g/L碘化鉀-0.5%氫氧化鉀。硒空心陰極燈196.0nm;主電流/輔電流120/60mA;負高壓350V;還原劑:3%硼氫化鉀-0.5%氫氧化鉀;載流10%的鹽酸。
1.5.2 選擇性測定-原子熒光光譜差減法測定銻硒價態
土壤浸提液按照以下儀器工作條件用原子熒光光譜儀測定進行測定,同時測定空白。
工作條件:泵速60r/min;載氣/屏蔽氣300/800mL/min;爐高8mm;主電流/輔電流80/40mA。還原劑:1.5%硼氫化鉀-0.5%氫氧化鈉;載流5%鹽酸。
(1)Sb(Ⅲ)和Sb(Ⅴ)的測定
用1.5.1節中Sb(Ⅲ)單標配制1、2、5、10、20 μg/L的Sb(Ⅲ)標準溶液,按樣品測定方法分別制作Sb(Ⅲ)和總Sb標準曲線。
銻空心陰極燈217.6nm;負高壓320V。
Sb(Ⅲ)的測定:取樣品5mL,加入0.2mol/L檸檬酸5mL,混勻,測定。
總Sb的測定:樣品5mL,加入3mL 3%硫脲-抗壞血酸和2mL鹽酸,混勻,放置30min,測定。
差減法計算Sb(Ⅴ)含量。
(2)Se(Ⅳ)和Se(Ⅵ)的測定
用1.2節中Se(Ⅳ)單標配制1、5、10、20、50 μg/L的Se(Ⅳ)標準溶液,按樣品測定方法分別制作Se(Ⅳ)和總Se標準曲線。硒空心陰極燈196.0nm;負高壓300V。
Se(Ⅳ)的測定:取樣品5mL,加入濃鹽酸1.25mL,混勻,測定。
總Se的測定:取樣品5mL,加入濃鹽酸2.5mL,沸水浴加熱30min,測定。
差減法計算Se(Ⅵ)含量。
1.5.3 陽離子交換樹脂分離-電感耦合等離子體質譜法測定鉻價態
土壤浸提液按照以下儀器工作條件用ICP-MS法進行測定,同時測定空白。ICP-MS儀器工作條件:射頻發生器功率1350W;冷卻氣流速13L/min;載氣流速0.78L/min;輔助氣流速0.8L/min;碰撞反應池(CCT)模式,碰撞氣(H2-He)流速2.0 mL/min;積分模式為峰面積;分析同位素為52Cr;Rh作為內標。
用1.2節中Cr(Ⅵ)單標分別配制1、5、10、100、500μg/L的Cr(Ⅵ)標準溶液。
分別取土壤浸提液5mL,從上面注入裝填好樹脂的離子交換柱,并用水淋洗,下口用25mL比色管收集流出液,定容至25mL,ICP-MS測定,計算土壤樣中Cr(Ⅵ)含量。另取未過柱的浸提液測定Cr總量,差減法計算Cr(Ⅲ)含量。
2 結果與討論
2.1 提取方法
目前土壤價態分析提取技術主要有振蕩、超聲、渦旋和微波萃取等,提取溫度大多在30~60℃之間[33-34,39]。為了防止過高溫度導致價態轉變和方便離心分離,本實驗用離心管作為浸提容器,參照采用50℃水浴振蕩提取30min的浸提方式,操作流程簡便。
2.2 測定方法的優化
價態分析色譜法最常用的兩種儀器是LC-ICP-MS和LC-AFS聯用儀。研究關注最多的是As元素,As在土壤中的含量高,污染嚴重,毒害性強,被國際癌癥研究機構(IARC)定為Ⅰ類致癌物質。土壤中三價砷As(Ⅲ)和五價砷As(Ⅴ)分別以亞砷酸鹽和砷酸鹽形式存在,As(Ⅲ)毒性大于As(Ⅴ),主要采用離子色譜-ICP-MS法[14-16]和LC-AFS法[17-19]測定。土壤中Cr(Ⅲ)一般以陽離子Cr3+形式存在,它是人體的必需微量元素,但劑量較高時有細胞毒性。Cr(Ⅵ)則是Ⅰ類致癌物質,pH>6時Cr(Ⅵ)以鉻酸根或重鉻酸根等陰離子形式存在;pH<6時Cr(Ⅵ)轉化為Cr(Ⅲ)。十多年前測定Cr價態采用AAS法較多,現在一般采用HPLC-ICPMS法[21-25],流動注射-AAS法[26]和AFS法[38]也有報道。Sb含量不高(《中國土壤背景值圖集》中Sb土壤背景值為0.38~2.98mg/kg),但毒性強,是2B類致癌物質。中國2018年出臺的《土壤環境質量農用地和建設用地土壤污染風險管控標準》中尚未設置Sb的限值,因此Sb受關注程度較低,價態研究較少。Sb(Ⅲ)和Sb(Ⅴ)分別以Sb3+離子和銻酸根形式存在,Sb(Ⅲ)的毒性大于Sb(Ⅴ),可用LCICP-MS法[27-28]或LC-AFS法[29-31]測定。近年硒的研究和應用受到廣泛關注,少量硒元素對人體有益,大量則有害。Se(Ⅳ)和Se(Ⅵ)分別以亞硒酸根和硒酸根形式存在,Se(Ⅳ)毒性大于Se(Ⅵ),主要采用HPLC-ICP-MS法[15,32-34]和LC-AFS法[18,35-36]測定。
2.2.1 液相色譜-原子熒光光譜聯用法測定砷銻硒價態
LC-AFS聯用法近年來在元素形態和價態測定的應用上得到認可[37-38],成為主要檢測方法之一。試液中的待測組分通過離子色譜柱分離后,進入氫化物發生-原子熒光光譜儀檢測。圖1是As、Sb、Se的LC-AFS聯用譜圖,兩種組分基線分離,峰形對稱無拖尾,響應值高,前處理簡便,無基質干擾,是目前理想的價態分析方法。本實驗首選采用LC-AFS聯用法測定砷銻硒價態。
(1)色譜柱的選擇

圖1 As、Sb、Se價態液相色譜-原子熒光譜圖Fig.1 LC-AFS spectra of valence states of As,Sb and Se
價態分離所采用的色譜柱一般分為兩大類。一類是C18柱或反相柱[18,21-22,32-33],只能分離有機物,使用時須添加離子對試劑如四丁基氫氧化銨(TBAH)、溴化四丁基銨(TBBA)等有機銨離子以及戊烷磺酸鈉、十二烷基磺酸鈉等烷基磺酸鹽。另一類是陰離子色譜柱[14-17,19,23-25,27-31,34-36],可分離無機陰離子,使用方便。本實驗比較了Dionex IonPac AS19陰離子色譜柱(250mm×4mm)和Hamilton PRP X-100陰離子交換柱(4.1mm×250mm,10μm)對幾種陰離子的分離效果,結果表明采用Dionex IonPac AS19陰離子色譜柱 (250mm ×4mm),峰形更對稱,分離效果更好。
(2)流動相及濃度的選擇
對于陰離子色譜柱,磷酸鹽緩沖液[37]較常用作As的流動相,如磷酸氫二鈉-磷酸二氫鉀[17]和磷酸氫二銨[18-19],還有的采用氫氧化鉀[14]、硝酸銨[15]、TMAH[16]等。本實驗對磷酸氫二鈉-磷酸二氫鉀和磷酸氫二銨進行試驗,二者都可行。本實驗分別配制了10、30、60、100、200mmol/L的磷酸氫二銨流動相,觀察保留時間、峰高、峰形等特性。為使兩峰基線分離,保留時間短,峰形對稱不拖尾,選用100mmol/L磷酸氫二銨(pH=6.0)作流動相。
文獻報道Sb的流動相有鄰苯二甲酸氫鉀-EDTA[27-28]、磷酸氫二銨-酒石酸[29]、甲醇-酒石酸銨[30]、EDTA[31]和磷酸鹽緩沖液[37]等。由于酒石酸對Sb有屏蔽作用[2,29],本實驗比較了鄰苯二甲酸氫鉀、磷酸氫二銨、磷酸二氫鉀等流動相,流動相中加入EDTA,使Sb3+離子與EDTA形成絡合物并在陰離子色譜柱上保留。試驗了幾種不同濃度,選擇最佳流動相為1mmol/L鄰苯二甲酸氫鉀-10mmol/L EDTA。
Se的流動相有檸檬酸[34]、磷酸二氫銨-甲醇[35]、檸檬酸銨[36]、磷酸鹽緩沖液[37]等。本實驗比較了磷酸氫二鈉-磷酸二氫鉀、磷酸氫二銨和碳酸氫銨等流動相,都可以使兩組分分離。為方便,采用與As相同的流動相100mmol/L磷酸氫二銨(pH =6.0),分離效果好。
(3)硼氫化鉀濃度的選擇
試驗了硼氫化鉀濃度在1% ~5%變化時As、Sb、Se價態峰高響應值的變化。選擇3%硼氫化鉀溶液,兩種價態都可以得到較高的峰高響應。
(4)載流鹽酸濃度的選擇
試驗了鹽酸濃度在3%~10%變化時As、Sb、Se價態峰高響應值的變化,確定5%、7%和10%鹽酸分別作為測定As、Sb、Se的載流。
(5)燈電流、負高壓、載氣流量的優化
隨著燈電流、負高壓增大,熒光強度也明顯增大,但不能無限提高燈電流和負高壓,否則會抬高基線,增大噪音。通過實驗,對于As、Sb、Se選定燈電流分別為80/40mA、90/45mA,120/60mA,負高壓分別為310V、350V、350V,基線平穩,信號響應值高。載氣在300mL/min時熒光強度最大,屏蔽氣流速在800~1000mL/min時熒光強度相差不多,1100 mL/min時熒光強度下降,故取儀器默認值載氣流速為300mL/min,屏蔽氣流速為900mL/min。
(6)紫外消解和還原劑碘化鉀濃度的選擇
Se(Ⅵ)不能與KBH4反應生成氫化物,但可以用還原的方法使Se(Ⅵ)轉變為Se(Ⅳ)[36]。本實驗發現在紫外消解開啟的情況下,Se(Ⅵ)可與碘化鉀在線反應還原成Se(Ⅳ),從而出現原子熒光響應信號。
研究了碘化鉀濃度(1、3、5、7、10g/L)對峰高響應的結果。結果表明,碘化鉀濃度變化對Se(Ⅳ)影響不大;而對Se(Ⅵ)有影響,不加碘化鉀時Se(Ⅵ)不出峰,碘化鉀濃度越高Se(Ⅵ)峰越高,5g/L以后趨于穩定。故選5g/L的碘化鉀溶液為在線還原劑。
2.2.2 離子色譜-電感耦合等離子體質譜聯用法測定砷鉻硒價態
LC-ICP-MS聯用法在近二十年前已開始大量應用于形態和價態研究,相比LC-AFS聯用法起步更早,檢出限更低[40-41],不需要柱前處理和氫化,多種元素如As、Se、Cr等可同時測定[15,18,42],但也有研究報道其檢出限比LC-AFS法高1~2個數量級[37],且價格昂貴,對有機溶劑耐受不好。
離子色譜屬于液相色譜的一種,本實驗通過離子色譜和ICP-MS之間的Peek管簡單連接,成功地對As、Cr、Se價態進行了分離。As、Se的液相分離條件與LC-AFS聯用法一致。Cr(Ⅲ)和Cr(Ⅵ)的測定采用HPLC-ICP-MS法較為理想。有研究者用C18柱,加入四丁基氫氧化銨(TBAH)與Cr(Ⅲ)形成離子對,乙酸銨-乙二胺四乙酸溶液等作為流動相[21];也有研究者用陰離子交換柱,乙二胺四乙酸二鈉(EDTA)絡合Cr(Ⅲ),硝酸銨作流動相[24-25],pH一般調節在7~9之間。經試驗,本實驗建立了如下測定Cr的方法:浸提液中加入0.38g的EDTA,水浴70℃加熱1h以絡合Cr3+離子,60mmol/L硝酸銨(pH=7.1)作為流動相,使它在Dionex IonPac AG7陰離子色譜柱上保留并與Cr(Ⅵ)分離,進入ICP-MS質譜儀檢測,使用內標Rh校正。
本實驗在離子色譜-ICP-MS聯用法測定Sb價態時未能成功,探究發現當流動相中含有EDTA時,有大量白色粉末狀固體在ICP-MS析出,堵塞霧化器噴嘴和整條廢液管,而無EDTA時則無此現象。已有研究報道六羥基銻酸鉀在堿性環境中會與鈉離子反應產生白色顆粒狀沉淀,引起色譜柱堵塞[39]。但同樣的流動相在LC-AFS聯用儀運行時卻未出現沉淀,原因有待研究。
實驗表明離子色譜-ICP-MS聯用法測定元素價態是可行的,干擾少,方法簡便,但本研究中該方法靈敏度不如LC-AFS法,如Se譜圖見圖2,基線噪聲明顯,檢出限比后者高一個數量級。是否存在儀器較老影響性能的問題,還有待探究。未購買專業接口和工作站軟件也影響了儀器使用的便利性,因此離子色譜-ICP-MS聯用法未作為首選。

圖2 100μg/L Se(Ⅳ)和Se(Ⅵ)的離子色譜-ICP-MS聯用譜圖Fig.2 IC-ICP-MS spectra of 100μg/L Se(Ⅳ)and Se(Ⅵ)
2.2.3 原子熒光光譜差減法測定銻硒價態
Sb、Se價態也可采用AFS差減法測定,其中Sb采用加掩蔽劑法。As因儀器原因暫無法測定,現有的AFS儀器無外在氫源,控制酸度法As(Ⅴ)要求的酸度過低,無法產生足夠的氫氣。文獻中應用較多的717陰離子交換樹脂吸附分離法在本次實驗中效果不理想,也未采用,還需繼續研究。
(1)銻的掩蔽劑和還原劑
離子交換態Sb提取液是0.2mol/L酒石酸,而酒石酸對Sb有掩蔽作用[2,29],導致LC-AFS聯用法測定的靈敏度低,宜采用AFS差減法。一份樣品加掩蔽劑過濾掉Sb(Ⅴ)的信號,達到測定Sb(Ⅲ)的目的。另一份樣品加還原劑,使Sb(Ⅴ)還原為Sb(Ⅲ),測定總Sb,差減法得到Sb(Ⅴ)含量。
常用作Sb(Ⅴ)掩蔽劑的有檸檬酸、檸檬酸鈉、氟化鈉、8-羥基喹啉、檸檬酸-氟化鈉、酒石酸[2,4-6]等。常用的Sb(Ⅴ)還原劑有碘化鉀、半胱氨酸和硫脲-抗壞血酸[2]。本實驗比較了檸檬酸、氟化鈉、檸檬酸-氟化鈉、酒石酸、碘化鉀、半胱胺酸、硫脲-抗壞血酸對等量Sb(Ⅲ)和Sb(Ⅴ)熒光強度的影響。圖3結果表明,酒石酸和氟化鈉對Sb(Ⅲ)和Sb(Ⅴ)都有掩蔽作用,信號僅為原來的三分之一。檸檬酸和檸檬酸-氟化鈉完全掩蔽了Sb(Ⅴ)的信號,并且檸檬酸對Sb(Ⅲ)還有增敏作用,故采用0.1mol/L檸檬酸作為掩蔽劑。碘化鉀、半胱胺酸和硫脲-抗壞血酸都使Sb(Ⅴ)還原為Sb(Ⅲ),并且增強了Sb(Ⅲ)的信號,其中硫脲-抗壞血酸信號強度最大,Sb(Ⅴ)還原完全,故采用硫脲-抗壞血酸作為還原劑。
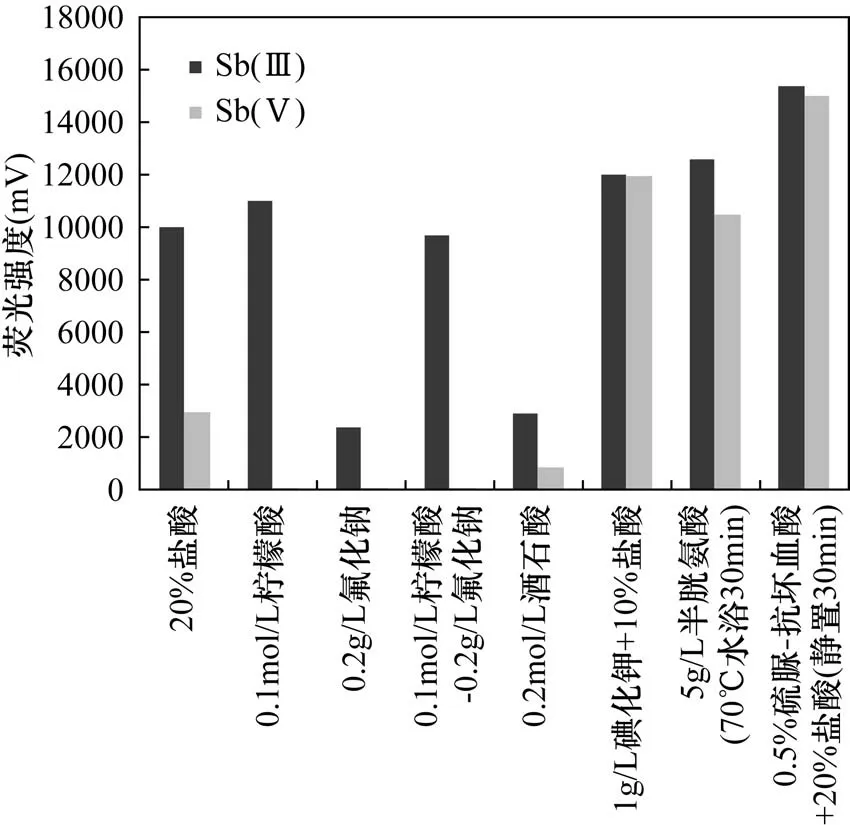
圖3 掩蔽劑和還原劑對等量Sb(Ⅲ)和Sb(Ⅴ)原子熒光強度的影響Fig.3 Effect of masking agent and reducing agent on the atomic fluorescence intensity of the same amount of Sb(Ⅲ)and Sb(Ⅴ)
(2)硒的還原條件
Se(Ⅵ)與KBH4不能形成氫化物,因此不會干擾Se(Ⅳ)的測定[7-8]。在加熱和高濃度鹽酸條件下Se(Ⅵ)還原成Se(Ⅳ),用AFS法檢測Se(Ⅵ)和總Se含量,差減法得到Se(Ⅵ)含量。
實驗比較了用碘化鉀、鹽酸、半胱氨酸等還原等量Se(Ⅳ)和Se(Ⅵ)后的熒光強度,結果如圖4所示。經多次條件試驗,碘化鉀和半胱氨酸無論是否加熱都屏蔽了Se(Ⅵ)和Se(Ⅳ),只有鹽酸沸水浴加熱還原后增強了Se(Ⅵ)的信號強度,與Se(Ⅳ)不加熱時相當,說明Se(Ⅵ)已全部轉化為Se(Ⅵ),故采用鹽酸作為還原劑。

圖4 還原劑對等量Se(Ⅳ)和Se(Ⅵ)原子熒光強度的影響Fig.4 Effect of reducing agent on atomic fluorescence intensity of the same amount of Se(Ⅳ)and Se(Ⅵ)
本實驗還考察了鹽酸濃度(20%、25%、30%、40%、50%、70%)和水浴溫度(50℃、70℃、90℃和100℃)對Se(Ⅵ)還原轉化率的影響。結果表明鹽酸濃度大于30%時Se(Ⅵ)轉化率最大,在沸水浴中Se(Ⅵ)完全轉化為Se(Ⅳ),從實驗方便考慮,選擇還原條件為33%鹽酸,采用100℃水浴加熱。
2.2.4 陽離子交換樹脂分離-電感耦合等離子體質譜法測定鉻價態
本實驗中Cr的價態采用了732陽離子交換樹脂分離,ICP-MS測定的方法。
(1)離子交換樹脂選擇
Cr3+是陽離子,可用732陽離子交換樹脂吸附,通過實驗觀察,經過動態離子交換柱吸附分離,在中性或弱堿性時Cr(Ⅲ)吸附率接近100%,而Cr(Ⅵ)幾乎不被吸附。收集流出液測定Cr(Ⅵ),原液測定總Cr,差減法計算Cr(Ⅲ)含量。因此實驗采用732陽離子交換樹脂對Cr(Ⅲ)和Cr(Ⅵ)進行分離。
(2)淋洗時間優化
選用的玻璃樹脂交換柱長25cm,容量5g,管徑較細,上樣淋洗時流速不快,接滿25mL流出液需30min左右。實驗表明,收集25mL洗脫液后,目標物Cr(Ⅲ)吸附率>99.5%,洗脫物Cr(Ⅵ)回收率>99.5%,淋洗完全。確定淋洗時間為30min。
(3)淋洗液用量優化
玻璃交換柱上分別加入1000μg/mL的Cr(Ⅲ)和Cr(Ⅵ)標準溶液1mL,分段收集淋洗流出液,每10mL一次,分析流出液中Cr含量。結果表明,收集25mL流出液后,Cr(Ⅵ)回收率>99.5%。
(4)質譜條件優化及干擾的消除
主要通過調節載氣、輔助氣、冷卻氣和碰撞氣的流速等工作參數,使進樣系統在一定的提升量下維持較高的霧化效率和足夠的信號強度。多原子干擾是ICP-MS測定過程中主要的干擾,鉻在m/z=52處常見的多原子離子干擾類型是40Ar12C+。本實驗采用碰撞反應池(CCT)技術,利用動能選擇的原理來消除干擾離子。
2.2.5 分析方法的線性范圍、檢出限、精密度和準確度
按規范要求進行檢出限、精密度和準確度實驗,結果見表1。各條標準曲線相關系數都大于0.999。文獻報道的AFS法或LC-AFS法的土壤檢出限在0.005~0.5μg/g,本實驗檢出限在0.007~0.020 μg/g,靈敏度高,滿足DD2005-03檢出限0.02μg/g的要求;精密度滿足RSD<15%的要求;準確度滿足加標回收率90.0%~110.0%的要求。

表1 土壤中As、Cr、Sb和Se價態方法線性范圍、檢出限、精密度(RSD)和加標回收率Table 1 Linear range,method detection limit,relative standard deviation and standard addition recovery of As,Cr,Sb and Se in soil samples
2.3 實際樣品測定
對采集的編號為N01~N20的土壤樣品,提取測定相應價態,并用消解法測定土壤中該元素總含量。表2結果表明,水溶態As含量較低,離子交換態As較高,其中As(Ⅴ)大大高于As(Ⅲ);土壤浸提液中Cr(Ⅵ)普遍高于Cr(Ⅲ),但土壤Cr總量高的樣品,浸提液中Cr含量并不高,沒有表現出相關性;土壤中Sb和Se含量本身都不高,水溶態和離子交換態的Sb和Se含量很低,在檢出限附近。
實驗還表明,As(Ⅲ)在中性和堿性條件下很不穩定,較易轉化為As(Ⅴ);在酸性條件下較穩定。Cr(Ⅵ)在酸性條件下很不穩定,易轉化為Cr(Ⅲ);在中性和堿性條件可以長期保存。Cr(Ⅲ)在酸性條件下穩定,標準宜儲存在鹽酸介質中。樣品處理時注意控制酸度,盡快測定。Sb(Ⅲ)在酸性條件下很不穩定,較易轉化為Sb(Ⅴ),能被土壤及二氧化硅完全吸附,水和酒石酸不能解脫,配制標準溶液須采用固體試劑,儲存時間不宜超過兩周。Se(Ⅵ)較不穩定,水溶液放置久了易部分轉化為Se(Ⅳ)。
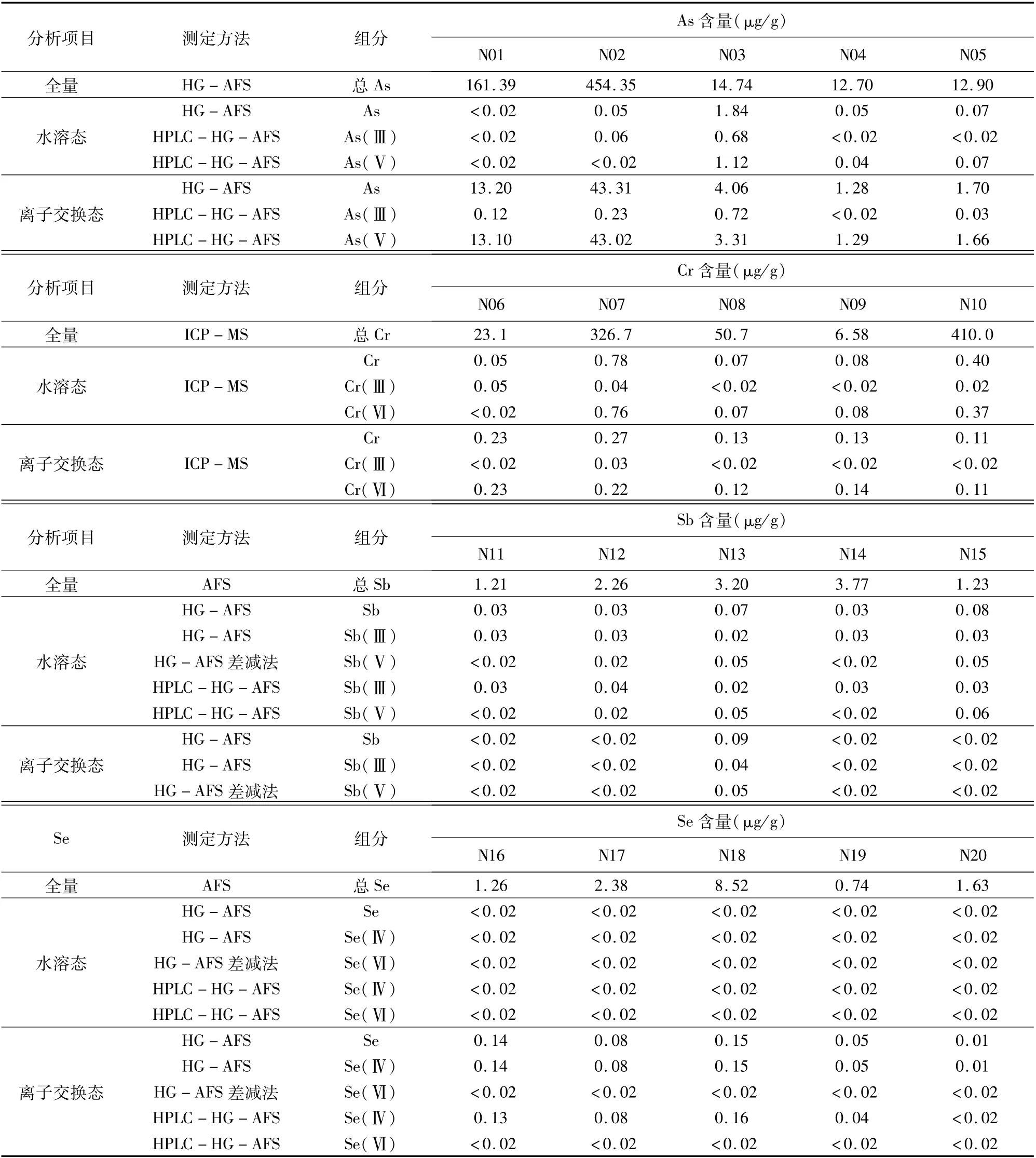
表2 實際土壤樣品中As、Cr、Sb和Se價態含量Table 2 Content of As,Cr,Sb and Se in actual soil samples
3 結論
本文建立了水浴振蕩加熱提取,液相色譜-原子熒光光譜聯用法測定As、Sb、Se價態的方法,與傳統非色譜法比較,簡化了前處理程序,排除了土壤樣品中的基質干擾,一次進樣同時測定兩個價態,提高檢測的準確度和靈敏度。Cr價態采用陽離子交換樹脂分離-電感耦合等離子體質譜法測定的方法,比石墨爐原子吸收光譜法的靈敏度高。為避免某些離子交換提取劑的屏蔽干擾作用,同時還建立了原子熒光光譜差減法選擇性測定Sb、Se價態的方法,儀器設備投入較低。對離子色譜-ICP-MS聯用法也進行了研究,因靈敏度較低并且使用中遇到的一些困難而未采用。
通過對實際土壤樣品的檢測,初步了解了土壤浸出液中As、Cr、Sb、Se價態存在現狀,價態浸出含量遠低于土壤總量,且未很好體現相關性。初步掌握了價態之間轉化規律,樣品采集、運輸、加工、儲存過程中必須注意酸堿度的控制,樣品處理后盡快測定。今后有必要對價態的轉化規律及測定進行深入探究;土壤離子交換提取液難以過濾澄清,給實驗的帶來的不便和相關影響還有待解決。
猜你喜歡
作文·小學低年級(2025年2期)2025-02-13 00:00:00
小雪花·小學生快樂作文(2024年11期)2024-12-31 00:00:00
作文·小學低年級(2024年2期)2024-04-29 00:00:00
作文·小學低年級(2023年3期)2023-04-29 00:00:00
小獼猴智力畫刊(2022年9期)2022-11-04 02:31:42
小主人報(2022年4期)2022-08-09 08:52:06
中學生數理化·中考版(2022年11期)2022-02-16 07:01:20
小哥白尼(趣味科學)(2019年6期)2019-10-10 01:01:50
發明與創新(2016年38期)2016-08-22 03:02:52
太空探索(2016年5期)2016-07-12 15:17:55
