AQP4-IgG陽性與AQP4-IgG陰性NMOSD的臨床學比較研究
2021-04-20 02:52:00武國良
中風與神經疾病雜志 2021年3期
武國良,彭 濤
視神經脊髓炎譜系疾病(NMOSD)是一種嚴重的中樞神經系統炎癥性脫髓鞘疾病,主要影響視神經和脊髓。據估計,每10萬人中約有0.5~10人患NMOSD,其中偏愛亞洲人和女性[1]。可根據核心臨床特征、血清水通道蛋白抗體及神經影像學特征進行診斷[2]。NMOSD患者表現為單發或復發性視神經炎、縱向廣泛的橫向脊髓炎和腦部病變,可導致嚴重殘疾甚至死亡。有60%~90%符合NMOSD診斷標準的患者可以檢測到AQP4-IgG,甚至有專家學者提出AQP4-IgG陰性的NMOSD是否存在。然而,一些NMOSD患者在急性期使用高敏感的細胞分析法(CBA)進行重復檢驗,AQP4-IgG仍然陰性。同時,AQP4-IgG陰性NMOSD患者在臨床發病特點上不同于AQP4-IgG陽性患者,具體機制目前尚不清楚。
1 資料和方法
1.1 研究對象 對2015年9月至2019年9月鄭州大學第一附屬醫院神經內科收治的345例 NMOSD患者進行回顧性分析。所有患者均處于急性期(初次發病或復發1 m內),并由至少兩位神經內科專家根據2015年NMOSD國際標準共同診治[2]。復發定義為新發癥狀持續24 h并有客觀的神經系統體征或新發的影像學改變。納入標準:(1)已知AQP4-IgG血清學狀態;(2)滿足2015年NMOSD國際診斷標準(見表1)。排除標準如下:(1)AQP4-IgG血清狀態未知;(2)隨訪數據缺失;(3)共存其他可能影響EDSS的神經或眼科疾病;(4)寡克隆免疫球蛋白(OCB)或副腫瘤相關抗體陽性;(5)共存其他可能影響診斷的疾病(如:淋巴瘤、脊髓血管病變、脊髓或腦部感染性病變等)。本研究經鄭州大學第一附屬醫院倫理委員會批準,獲得患者的知情同意。

表1 2015視神經脊髓炎譜系疾病(NMOSD)診斷標準
1.2 研究方法 記錄所有入組病例的人口學、臨床表現、血液學化驗、腦脊液化驗及磁共振影像學資料等。所有患者使用基于轉染細胞分析法(cell based assays,CBA)或間接免疫熒光法行血AQP4抗體檢測,部分患者同時行血MOG-IgG檢測。大部分患者同時送檢血樣檢測IgG寡克隆區帶(OCB)、抗核抗體、同型半胱氨酸、維生素B12、血沉、C反應蛋白及血常規等。同時大部分患者在入院后接受腰穿檢查,送檢腦脊液常規、生化、細胞學、OCB。采用3.0T MRI進行頭及脊髓MRI檢查,成像序列包括T1加權像、T2加權像、液體衰減反轉恢復序列(FLAIR)、彌散加權成像(DWI)。MRI檢查結果由放射科專家與神經內科專家一起評定。
2 結 果
2.1 一般資料 共有345例NMOSD患者,AQP4-IgG陽性組244例(71%),AQP4-IgG陰性組101例(29%);陽性組與陰性組分別有39和26例患者送檢MOG-IgG,其中陰性組出現2例MOG-IgG陽性患者。共有女性279例(81%),其中陽性組214例,陰性組65例;男性66例(19%),陽性組30例,陰性組36例。陽性組女性發病率明顯高于AQP4-IgG陰性組(P=0.000)。起病年齡10-82歲不等,平均起病年齡(41.20±16.60),陽性組(42.15±15.78),陰性組(38.93±18.31)。陽性組平均起病年齡高于陰性組,但組間差異不顯著(P=0.101)。AQP4-IgG陽性與AQP4-IgG陰性NMOSD患者臨床特征見表2。
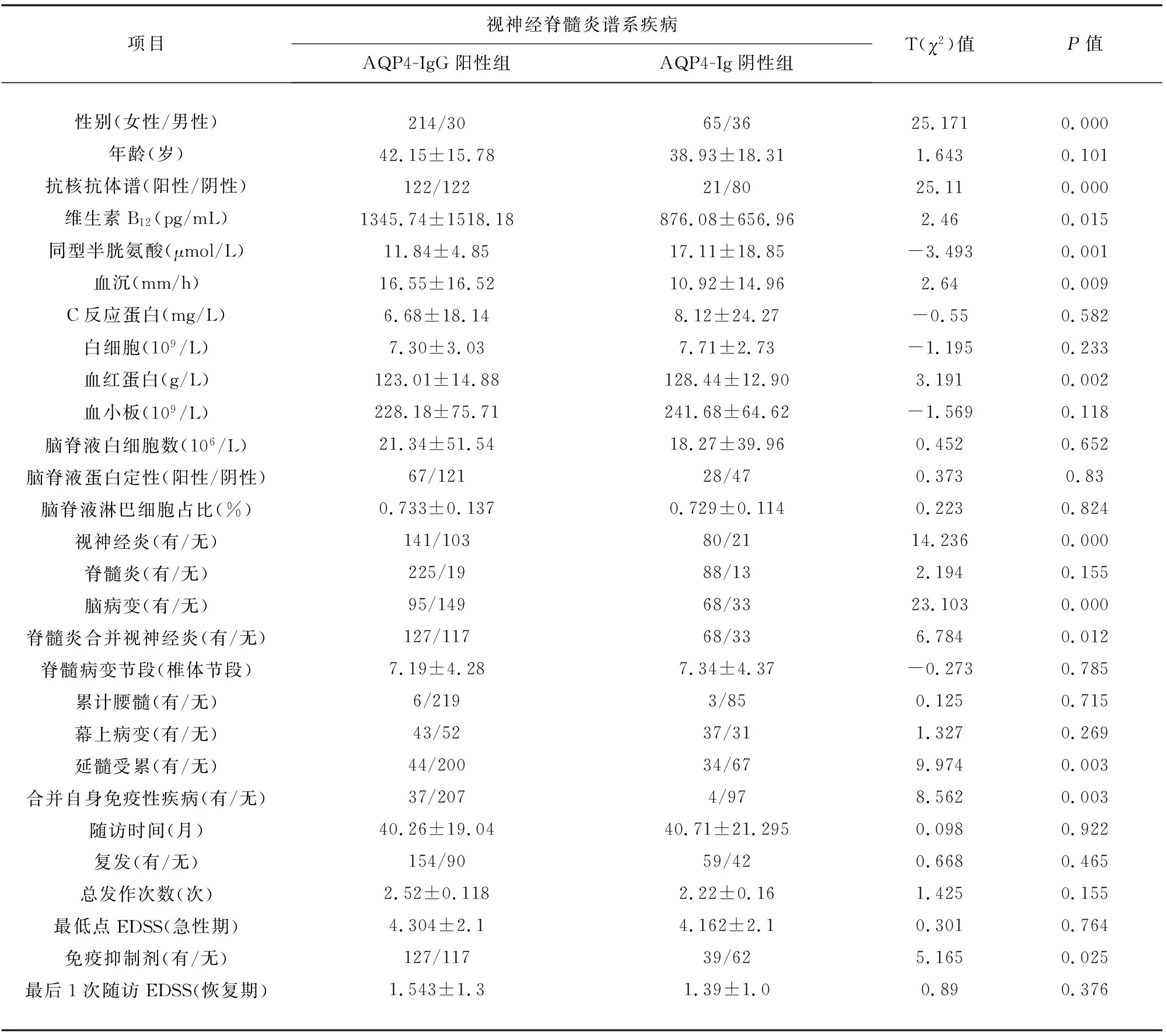
表2 兩組NMOSD患者的臨床特點比較
2.2 臨床特點 臨床疾病進程中有脊髓炎發作者313例(91%),陽性組225例,陰性組88例;有視神經炎者221例(64%),陽性組141例,陰性組80例;有腦部病變者163例(47%),陽性組95例,陰性組68例;既有視神經炎又有脊髓炎者195例(57%),陽性組127例,陰性組68例。AQP4-IgG陰性組的腦部病變、視神經炎及既有視神經炎又有脊髓炎的發病率明顯高于陽性組(P=0.000;P=0.000;P=0.012),脊髓炎發病率在兩組間無統計學差異(P>0.05)。
2.3 MRI影像學特點 脊髓病變常累計頸胸髓,且常同時受累,腰髓極少受累。脊髓病變節段數介于1~20不等,以長節段病變為主,可不連續;平均脊髓病變節段數(7.23±4.30),陽性組(7.19±4.28),陰性組(7.34±4.37);共有9例患者累計腰髓,陽性組6例,陰性組3例,脊髓病變節段及腰髓受累在兩組間無統計學意義(P>0.05)。腦部病灶形態多樣,多呈斑片狀、點片狀分布,邊界清晰或模糊,呈長T2信號,部分病灶可呈斑點狀、條片狀強化。延髓是最常見的受累部位,腦干、大腦皮質、丘腦、橋臂、基底節、小腦半球、側腦室旁白質、胼胝體、大腦腳、半卵圓中心等部位也可受累。共有78例患者累計延髓,陽性組44例,陰性組34例,AQP4-IgG陰性組延髓受累率明顯高于陽性組(P=0.003)。幕上病變患者80例,陽性組43例,陰性組37例,兩組間差距不明顯(P>0.05)。
2.4 血及腦脊液化驗結果 抗核抗體譜陽性患者143例(41%),陽性組122例,陰性組21例;AQP4-IgG 陽性組抗核抗體譜陽性率明顯高于陰性組(P=0.000)。平均維生素B12濃度(1140.39±719.80),陽性組(1345.74±1518.18),陰性組(876.08±656.96);平均同型半胱氨酸濃度(13.30±10.80),陽性組(11.84±4.85),陰性組(17.11±18.85);平均血沉(15.05±16.29),陽性組(16.55±16.52),陰性組(10.92±14.96)。平均C反應蛋白濃度(6.96±19.94),陽性組(6.68±18.14),陰性組(8.12±24.27)。AQP4-IgG陽性組維生素B12、血沉明顯高于陰性組(P=0.015;P=0.009),AQP4-IgG陰性組同型半胱氨酸明顯高于陽性組(P=0.001),C反應蛋白在兩組間無統計學意義(P>0.05)。平均白細胞計數(7.42±2.95),陽性組(7.30±3.03),陰性組(7.71±2.73);平均血紅蛋白濃度(124.62±14.52),陽性組(123.01±14.88),陰性組(128.44±12.90);平均血小板計數(232.17±72.78),陽性組(228.18±75.71),陰性組(241.68±64.62)。AQP4-IgG陰性組血紅蛋白值明顯高于陽性組(P=0.002),白細胞及血小板計數在兩組間差異不大(均P>0.05)。腦脊液白細胞計數(19.97±47.98),陽性組(21.34±51.54),陰性組(18.27±39.96);腦脊液淋巴細胞百分數(0.73±0.13),陽性組(0.733±0.137),陰性組(0.729±0.114);腦脊液蛋白定性陽性患者95例,陽性組67例,陰性組28例。腦脊液中的白細胞數、淋巴細胞比率及蛋白定性在AQP4-IgG 陽性與陰性組間均無統計學差異(均P>0.05)。
2.5 共患其他自身免疫性疾病 共患其他自身免性疫疾病患者41例(12%),陽性組37例,陰性組4例。AQP4-IgG 陽性組其他自身免疫性疾病共患率明顯高于陰性組(P=0.003)。
2.6 治療及轉歸情況 急性期兩組患者均采用大劑量激素沖擊治療,對激素不敏感患者加用免疫球蛋白治療,效果可;緩解期兩組患者均口服小劑量潑尼松;共有166例(47%)患者使用免疫抑制劑,陽性組127例,陰性組39例;陽性組免疫抑制劑使用率明顯高于陰性組(P=0.025)。總隨訪時間(40.45±19.90)月,陽性組(40.26±19.04),陰性組(40.71±21.295);隨訪時間在兩組間差距不大(P>0.05)。隨訪時間內共有213例(62%)患者復發,陽性組154例,陰性組59例;總發作次數(2.43±1.77),陽性組(2.52±1.84),陰性組(2.22±1.60)。復發例數及總發作次數在兩組間無差距(P>0.05)。最低點EDSS評分(4.244±2.08),陽性組(4.304±2.1),陰性組(4.162±2.1);最后1次隨訪時EDSS評分(1.444±1.16),陽性組(1.543±1.3),陰性組(1.39±1.0);兩次EDSS評分在兩組間均無統計學意義(均P>0.05)。
3 討 論
NMO最初被稱為毀滅性的視神經脊髓炎癥性脫髓鞘疾病,是指同一患者視神經炎和脊髓炎的共同發生。2004年發現了AQP4-IgG這一種特定的疾病生物標志物,從而對這種疾病有了新的認識,該生物標志物最初被稱為“NMO-免疫球蛋白G”(NMO-IgG),第二年確定了其靶抗原水通道蛋白-4(AQP4)。隨后AQP4-IgG被正式納入2006年診斷標準[3],盡管NMO的診斷仍然需要視神經炎和脊髓炎這兩個條件。術語“NMOSD”首次被引入是在2007年[4],當時發現AQP4-IgG與NMO相關。2015年國際專家組(IPND)在診斷標準中提出了整個臨床綜合征的術語“NMOSD”,并將該疾病分為AQP4-IgG陽性NMOSD和AQP4-IgG陰性NMOSD(或未知血清狀態)[2]。但現已有多項研究證實了AQP4-IgG陰性與陽性NMOSD患者存在很多差異[5,6]。
在我們的研究中,女性與男性發病率之比約4∶1,陽性組約7∶1,陰性組約2∶1。兩組的女性發病率都明顯高于男性,但陽性組的女性發病率顯著高于陰性組。這與其他研究相一致[7,8]。陽性組與陰性組的平均發病年齡分別為42和39歲,陽性組的起病年齡稍大于陰性組,但差異不明顯[8]。另一項研究發現AQP4-IgG陰性患者發病年齡較小,女性比例較高[9]。
我們發現脊髓炎是最常見的臨床發作形式,視神經炎及腦部病變次之。91%的NMOSD患者經歷過至少1次橫貫性脊髓炎發作,陽性組92%,陰性組87%;64%的NMOSD患者有視神經炎,陽性組與陰性組分別有58%和80%;47%的NMOSD患者有腦部病變,陽性組39%,陰性組67%;57%的NMOSD患者或同時或先后經歷過脊髓炎和視神經發作,其中陽性組52%,陰性組67%。與陽性組相比,陰性組患者更容易出現視神經炎、腦部病變及脊髓炎和視神經炎[10]。其他研究亦發現血清陰性組同時發生視神經炎和脊髓炎更為常見[8,11]。另一項關于AQP4-IgG陽性與陰性NMOSD患者長期MRI表現的研究[12]發現血清陰性組有更多患者在起病時發生腦損害,血清陽性組患者隨著時間的推移容易發生腦損害,之后隨訪時兩組間差異消失。廣泛腦病變的存在可能預示著更高的疾病活動性和更差的預后,在NMOSD患者中,全身炎癥可能是廣泛腦病變形成的關鍵因素[13]。
NMOSD常表現為長節段橫貫性脊髓炎(LETM),多累及頸胸髓,腰髓受累較為少見,軸位上病灶多位于脊髓中央。我們發現陽性組與陰性組的腰髓受累率及脊髓病變節段數相似[10]。但我們在以前的研究中注意到血清陽性患者脊髓受累節段數更多[8,12],未來需要其他研究來驗證。腦部病灶形態多樣,多呈斑片狀、點片狀分布,邊界清晰或模糊,呈長T2信號,部分病灶可呈斑點狀、條片狀強化。延髓是最常見的受累部位,腦干、大腦皮質、丘腦、橋臂、基底節、小腦半球、側腦室旁白質、胼胝體、大腦腳、半卵圓中心等部位也可受累。延髓受累在陰性組更常見,但惡心嘔吐等極后區綜合征較少見;幕上病變在兩組間無明顯差距。其他研究發現陽性與陰性患者在幕上腦病變、腦干病變方面無統計學差異[8]。
在我們的研究中,143例NMOSD患者抗核抗體譜陽性,在內分泌科專家的協助診斷下,41例患者滿足SS/SLE等常見自身免疫性結締組織病的診斷標準,陽性組37例,陰性組3例。其中干燥綜合征最常見,系統性紅斑狼瘡次之,類風濕性關節炎、甲亢、重癥肌無力、強直性脊柱炎等相對少見。我們的結果與既往研究一致,AQP4-IgG陽性NMOSD患者常與SS/SLE或非器官特異性自身抗體并存,但其是一個獨立的疾病實體,而不是SS/SLE的血管病變或其他并發癥[14]。對中國NMOSD及MS患者進行抗核抗體譜篩查,NMOSD患者的血清陽性率明顯高于MS患者,抗核抗體和AQP4-IgG顯著提高了NMOSD診斷試驗的敏感性,而不影響特異性,并可能促進早期干預[15]。
AQP4-IgG陰性組的同型半胱氨酸(Hcy)及血紅蛋白值明顯高于陽性組,陽性組的維生素B12及血沉值明顯高于陰性組;同時,陽性組的Hcy及陰性組維生素B12均值都高于正常值范圍,兩組的血沉及血紅蛋白值均在正常值范圍。目前關于Hcy對免疫系統影響的研究較少,但Hcy與T、B淋巴細胞等免疫細胞的相關性是明確的[16,17]。Hcy可通過多條炎癥通路損傷血管內皮細胞,破壞血腦屏障,還可以影響內皮細胞釋放細胞因子,激活或募集中性粒細胞等炎性細胞到達局部,進一步加重對血腦屏障的損傷[18]。因此,高同型半胱氨酸(Hhcy)可能通過上述機制,使炎性細胞及炎性因子大量進入中樞,對神經細胞造成損傷。有研究發現HHcy與阿爾茨海默病、帕金森病等神經變性疾病相關,但具體機制仍不是很清楚[19,20]。此外,血漿Hcy水平升高不僅在MS患者中觀察到,而且與MS的進展有關[21]。其他研究發現EDSS評分≥4的急性期NMOSD患者Hcy和ESR水平顯著升高[22]。維生素 B12可能通過參與“葉酸循環”而對Hcy的代謝起著關鍵作用。VitB12含量不足時Hcy無法繼續合成甲硫氨酸,導致大量Hcy蓄積于血液之中,因而推測VitB12可能通過調節Hcy的代謝而影響炎癥過程。故維生素B12及同型半胱氨酸可能在NMOSD患者的發病過程中發揮一定的作用。有研究發現在伴有廣泛腦損害的NMOSD患者中,平均EDSS評分與ESR存在顯著正相關,這一結果提示了炎癥與疾病活動和NMOSD臨床殘疾之間的潛在聯系[13,22]。與陰性組相比,陽性組血紅蛋白值較低可能與血液系統免疫性疾病有關,也可能是由較低的維生素B12造成的。陽性組有1例患者合并自身免疫溶血性貧血,曾有過關于NMOSD合并自身免疫性溶血的個案報導[23]。此外,腦脊液白細胞、淋巴細胞百分數、蛋白定性在兩組間差距不明顯[8]。
2011年Mader等人第一次報告發表了AQP4-IgG血清陰性NMOSD患者中發現MOG-IgG介導的免疫應答;而在AQP4-IgG介導的體液免疫應答中,MOG不是目標抗原。現已有多項研究發現MOG-IgG陽性與AQP4-IgG陽性NMOSD在發病機制、臨床發病特點及治療反應方面均存在很大的差異。病理上,AQP4-IgG陽性NMOSD是一種自身免疫性星形細胞病變,而MOG-IgG陽性NMOSD是一種炎癥性脫髓鞘疾病。大約1/3至1/4的AQP4-IgG血清陰性NMOSD患者存MOG-IgG[24],但我們僅在7%的AQP4-IgG陰性NMOSD患者中發現MOG-IgG。MOG-IgG陽性NMOSD有較高比例的男性,較年輕的發病年齡,多為白種人,以及在影像學上更有可能累及圓錐和深灰質結構。他們更多的表現為視神經炎和橫向脊髓炎,多為雙側同時發生視神經炎,單相病程較常見,預后更好[25]。
所有患者在急性期采用大劑量激素或免疫球蛋白治療后效果可,我們發現兩組患者急性發作期癥狀最重時的殘疾程度(EDSS評分)相當;緩解期47%的患者使用免疫抑制劑控制病情進展,陽性組患者的免疫抑制劑使用率明顯高于陰性組,硫唑嘌呤最常用,嗎替麥考酚酯分散片次之,環磷酰胺、他克莫司、利妥昔單抗相對少用。經過40個月左右的隨訪后,62%的患者復發,但復發患者例數、總發作次數及最后1次隨訪時的EDSS評分在兩組間差距不明顯[10,26]。其他研究發現血清陰性患者更常見的是單相病程,陽性患者更常見復發[8]。這可能跟我們陰性組患者的免疫抑制劑使用率低有關。
我們的研究有一些局限性:首先,這是一項回顧性研究,我們的數據是基于我們醫院的病歷記錄。其次,只有小部分患者進行了MOG抗體檢測,所以我們的數據不足以分析MOG抗體陽性患者亞群。最后,由于是單中心研究,結果可能不能推廣到其他人種。未來需要進一步的設計研究來更加詳細具體的探討NMOSD及各亞組患者的疾病特征。
4 結 論
雖然2015版NMOSD的診斷標準把血清AQP4-IgG陽性與陰性患者都歸為NMOSD,但兩組患者的臨床疾病特征還是存在差異的。AQP4-IgG陽性組女性發病率、血清抗核抗體譜陽性率及其他自身免疫性疾病共患率更高,其維生素B12及血沉值也高于陽性組;AQP4-IgG陰性組更常見視神經炎、腦病變及既有視神經炎又有脊髓炎,其同型半胱氨酸及血紅蛋白值更高。隨著MOG-IgG在AQP4-IgG陰性NMOSD患者中的出現,多項研究證實了其與AQP4-IgG陽性患者在發病機制、臨床發病特點方面存在差異,使人們開始意識到NMOSD患者可能應該被重新分類。但是關于陰性NMOSD患者的發病機制、臨床特點、治療及其預防復發等仍存在很多未知,需要進一步的研究。
猜你喜歡
體育科技文獻通報(2022年3期)2022-05-23 13:46:54
中老年保健(2021年3期)2021-08-22 06:50:04
天津外國語大學學報(2021年3期)2021-08-13 08:32:18
遼金歷史與考古(2021年0期)2021-07-29 01:06:54
昆明醫科大學學報(2021年1期)2021-02-07 01:06:36
現代臨床醫學(2021年1期)2021-01-26 00:56:02
中華養生保健(2020年4期)2020-11-16 01:31:40
中西醫結合肝病雜志(2020年2期)2020-10-27 02:18:50
科技傳播(2019年22期)2020-01-14 03:06:54
民用飛機設計與研究(2019年4期)2019-05-21 07:21:24
