脂肪酶催化合成對映純硫雜環(huán)戊烷動力學模型
2021-04-02 12:06:41唐慧王繁業(yè)
青島大學學報(工程技術(shù)版) 2021年1期
唐慧 王繁業(yè)


摘要:? 為有效提高醫(yī)藥中間體對映純1,3-氧雜硫雜環(huán)戊烷(PR)的產(chǎn)率,本研究對動態(tài)動力學拆分,制備PR,并建立其動力學模型。采用一鍋法合成對映純((R)-5乙酰氧基-1,3-氧雜噻喃-2-基)乙基苯甲酸酯(PR),其中,催化劑為固定化絲孢酵母脂肪酶(Trichosporonlaibachii CBS 5791),底物為2-(苯基甲氧基)乙醛(A),1,4-二硫-2,5-二醇(B)和乙酸苯酯(D)。研究結(jié)果表明,其轉(zhuǎn)化率可以達到99.6%,產(chǎn)率為97.3%,對映體過量百分率(enantiomeric excess,ee%)為96.5%,所建立的反應體系動力學模型,很好地擬合了實驗數(shù)據(jù),表明可逆半硫代乙縮醛轉(zhuǎn)化反應動力學遵從冪律方程,而酶催化內(nèi)酯化反應為帶產(chǎn)物抑制的序列機制。該研究可高效合成PR,最大限度地提高了生產(chǎn)效率。
關(guān)鍵詞:? 動力學模型;? 脂肪酶;? 半硫縮醛轉(zhuǎn)化;? 1,3-氧硫雜環(huán)戊烷;? 酶促對映選擇性合成
中圖分類號: TQ463+.21; O643.32文獻標識碼: A
作者簡介: ?唐慧(1995-),女,碩士研究生,主要研究方向為藥物合成。
通信作者: ?王繁業(yè)(1965-),男,博士,教授,主要研究方向為制藥工程和藥物化學。? Email: fywang8209@163.com
手性1,3-氧雜硫雜環(huán)戊烷是許多藥物的重要中間體,如拉米夫定是用于治療HIV感染和慢性乙型肝炎最有效的藥物之一[1]。酶促不對稱合成或拆分制備對映體純1,3-氧雜硫雜環(huán)戊烷,由于其良好的立體選擇性,高效、溫和的反應條件和環(huán)境友好性而受到廣泛關(guān)注[24]。Ren Y等人[1]通過多酶級聯(lián)的方法,使用表面活性劑處理枯草桿菌蛋白酶嘉士伯和南極假絲酵母脂肪酶B,制備對映體(2R,5R)-1,3-氧雜硫雜環(huán)戊烷,使ee%>99%;Chen Y等人[5]通過產(chǎn)酸克雷伯菌介導的全細胞催化制備對映體富集的拉米夫定前體;Hu L等人[6]提出了加成-環(huán)化-乙酰化反應,采用新型表面活性劑處理的枯草桿菌蛋白酶,催化動態(tài)動力學拆分制備1,3-氧雜硫雜環(huán)戊烷。近年來,動態(tài)動力學拆分已在許多復雜的拆分系統(tǒng)中使用[69],但目前酶法合成手性1,3-氧雜硫雜環(huán)戊烷中間體仍無法達到滿意的對映體純度和產(chǎn)率。通過不同酶可以獲得不同構(gòu)型的對映體,例如,枯草桿菌蛋白酶通過催化拉米夫定前體進行不對稱合成生成拉米夫定,而南極假絲酵母的脂肪酶B、洋蔥伯克霍爾德氏菌和熒光假單胞菌的脂肪酶催化結(jié)果則與拉米夫定優(yōu)勢構(gòu)型相反[2,10]。T. laibachii已成功用于酮戊芬[11]的拆分和ε-己內(nèi)酯的原位開環(huán)聚合反應[12]。基于此,本文建立了脂肪酶催化合成對映純硫雜環(huán)戊烷動力學模型,該反應是加成-環(huán)化-乙酰化反應[6],反應中需要考慮不同組分之間存在的相互作用[13],可能的反應機制包括乒乓(Bi-Bi)機制[14]或三元復合機制(有序和隨機順序Bi-Bi)[15]。分析結(jié)果表明,本研究所建立的動力學模型,可揭示手性1,3-氧雜硫雜環(huán)戊烷的反應機理。該研究對提高反應速率具有重要意義。
1實驗部分
1.1實驗材料及分析測試/實驗材料和儀器
2-(苯基甲氧基)乙醛(99%),1,4-二硫-2,5-二醇(98%)和乙酸苯酯(99%)購于阿拉丁公司。以比活力為1 280 U/g的交聯(lián)固定化T. laibacchiii CBS5791脂肪酶為催化劑,使用前將催化劑真空干燥2 d。采用原位固定化方法制備了交聯(lián)固定化T. laibacchii CBS5791脂肪酶[16]。對于((R)-5-乙酰氧基-1,3-氧雜硫雜環(huán)戊烷-2-基)苯甲酸乙酯(PR)和((S)-5-乙酰氧基-1,3-氧雜硫雜環(huán)戊烷-2-基)苯甲酸乙酯(PS),采用高效液相色譜(High-Performance Liquid Chromatography,HPLC)分析,HPLC為Agilent RRLC 1200,色譜柱保持在30 ℃,檢測波長為210 nm。采用Chiracel OJ-H柱(4.6 mm×250 mm),進樣體積為5 μL,流動相為正己烷∶2-丙醇(即體積比為92∶8),流速為0.8 mL/min。采用氣相色譜法(Gas Chromatography,GC)分析2-(苯基甲氧基)乙醛和苯酚的濃度,SHIMADZU GC-2010 plus的毛細管柱為HP-FFAP(Agilent,內(nèi)徑30 mm×0.25 mm,薄膜0.25 μm)。通過程序升溫,首先將柱溫150 ℃維持在2 min,以10 ℃/min升至200 ℃,并保持1 min,以10 ℃/min升至270 ℃,然后保持15 min。將氮氣作為載氣,在分流模式下,噴射器溫度為250 ℃,分流比為20,火焰離子化檢測儀(Flame Ionization Detector,F(xiàn)ID)的溫度保持在270 ℃。
1.2酶法合成((R)-5-乙酰氧基-1,3-氧雜硫雜環(huán)戊-2-基)苯甲酸乙酯
固定化絲孢酵母脂肪酶催化的一鍋法反應步驟如下,將10 mL反應瓶置于水浴搖床,搖床轉(zhuǎn)速50~250 r/min,反應溫度40 ℃,反應時間48 h。反應體系中,2-(苯基甲氧基)乙醛的濃度為1.6 mmol/L,1,4-二硫-2,5-二醇的濃度為0.8 mmol/L,三乙胺的濃度為0.65 mmol/L,乙酸苯酯的濃度為3.55 mmol/L和四氫呋喃的體積為5 mL,其初始水的質(zhì)量分數(shù)為0.46%。加入酶引發(fā)反應,酶濃度為1~6 U/mL。反應結(jié)束后,過濾分離脂肪酶,將30 mL乙酸乙酯加入濾液,用飽和碳酸氫鈉溶液洗滌。將洗滌的濾液離心分離為水相和有機相,水相用乙酸乙酯(30 mL)萃取兩次,最后將合并的有機相用飽和氯化鈉洗滌,洗滌的有機相經(jīng)硫酸鎂干燥。真空除去溶劑,并將粗產(chǎn)物使用柱色譜法純化(己烷∶乙酸乙酯=6.5∶1)。
1.3內(nèi)、外擴散實驗
使用不同粒徑的脂肪酶進行內(nèi)擴散實驗,以研究脂肪酶的粒徑對化學-酶系統(tǒng)反應速率的影響。酶的粒徑為250~850 μm,搖床轉(zhuǎn)速為50~250 r/min,進行了外擴散實驗,以研究轉(zhuǎn)速對PR的影響。在內(nèi)、外擴散實驗中,除了反應時間為6.0 h外,其他反應條件與1.2相同。
2模型
2.1反應機理和模型
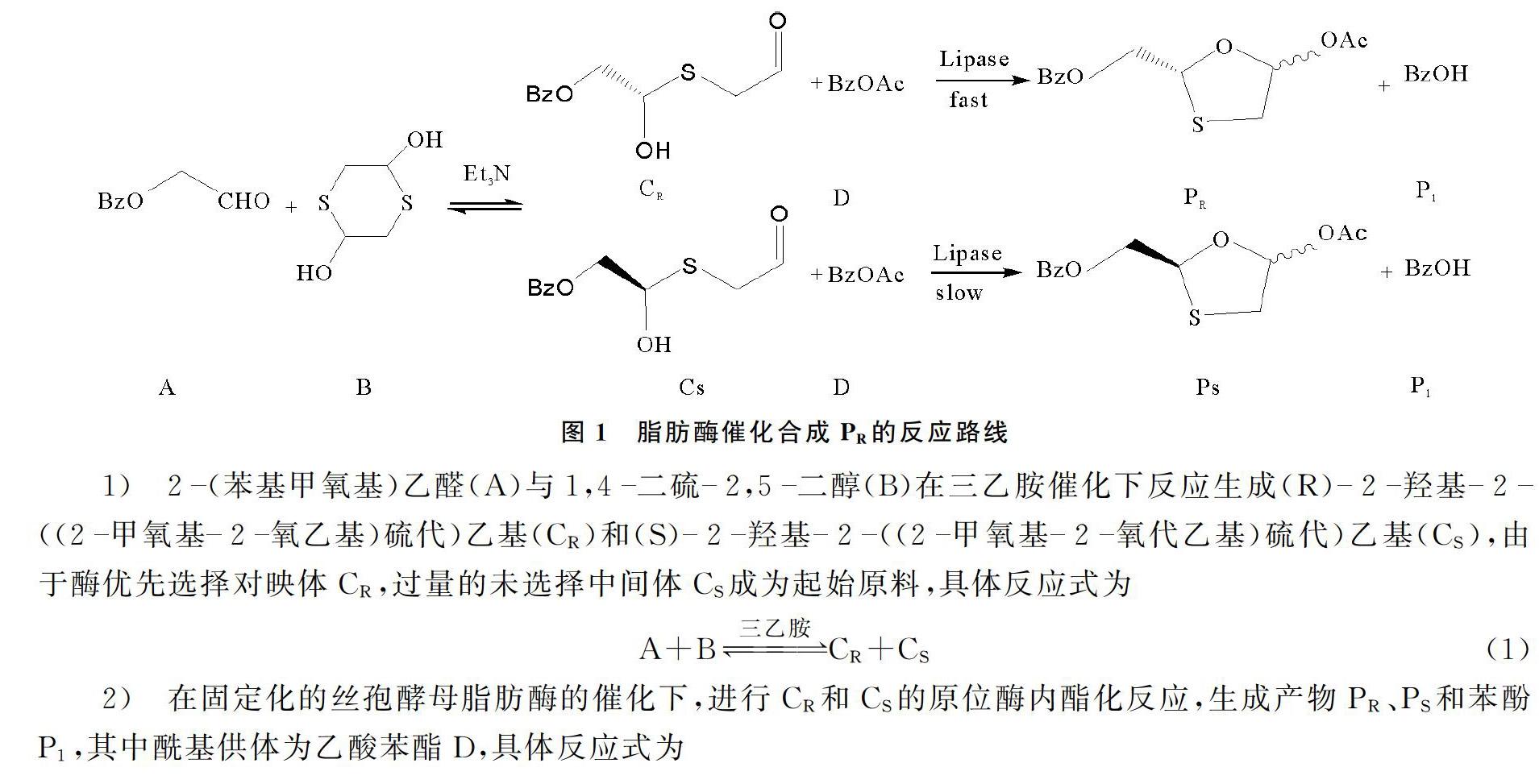
脂肪酶催化合成PR的反應路線如圖1所示[11],這是一個動態(tài)動力學拆分體系,包含3個反應,其中一個是可逆的半硫代縮醛轉(zhuǎn)化,兩個是脂肪酶催化的內(nèi)酯化反應,生成兩個對映體。
對動力學模型進行假設,生成最終產(chǎn)物PR,PS和P1的最后一步是不可逆反應,所建立的動力學模型被認為是固有的反應動力學模型,這一假設將在隨后的內(nèi)部和外部質(zhì)量擴散實驗中得到驗證;在整個反應過程中,酶活性的損失可以忽略不計。本研究反應后固定化脂肪酶的剩余活力大于95.4%,反應體系溫和[1718],并沒有像過氧化物或者酸性物質(zhì)那樣導致酶的失活。該反應中,一種酶同時催化反應式(2)和式(3),具有相同的脂肪酶活性位點,兩個平行反應交織在一起。反應式(2)和式(3)中沒有離去基團不遵循Bi-Bi機制,離去基團是Bi-Bi機制的特征之一[14]。因此,本研究將考慮順序機制[15]。CR和CS底物的濃度應近似,因為反應式(2)和式(3)是可逆的半硫縮醛轉(zhuǎn)化。因此,在建立動力學方程式時,CR和CS并未被作為兩個真正獨立的底物處理,在建立反應動力學模型時,可減少動力學模型參數(shù)。
反應式(1)采用帶有可逆項的冪律方程作為動力學方程。反應式(2)給出了序列反應機制,用于脂肪酶催化的環(huán)化和酯化序列反應機制如圖2所示。首先,底物D與脂肪酶E結(jié)合形成ED,ED與CR結(jié)合,產(chǎn)生脂肪酶三元復合物EDCR,將EDCR轉(zhuǎn)換為EP1PR,產(chǎn)物PR從EP1PR釋放,剩下EP1。最后EP1釋放產(chǎn)物P1,酶E再轉(zhuǎn)化為原始形式。反應式(3)與反應式(2)類似。式中,k1~k6和k-1~k-6為速度常數(shù)項與底物/產(chǎn)物濃度項的乘積。
3結(jié)果和討論
3.1內(nèi)、外擴散的影響
由于使用了多孔載體的固定化脂肪酶,因此有必要進行內(nèi)擴散實驗。A,B和D初始摩爾濃度分別為0.288,0.144和0.64 mmol/L,固定化脂肪酶的濃度為6.0 U/mL。當溫度為40 ℃,攪拌速度為150 r/min,給出不同固定化脂肪酶粒徑的實驗結(jié)果,固定化酶粒徑對PR產(chǎn)率的影響如圖3所示。由圖3可知,隨著固定化酶粒徑從0.5 mm增加到0.85 mm,PR的產(chǎn)率從46.1%逐漸降低至28.1%,說明在此粒徑范圍內(nèi),粒徑的增加與PR的產(chǎn)率成反比,酶催化受擴散限制。因此,如果粒徑大于0.5 mm,內(nèi)擴散對PR產(chǎn)率的影響較大。由圖3還可以看出,當珠粒徑在0.25~0.5 mm之間變化時,內(nèi)部傳質(zhì)對PR產(chǎn)率的影響可以忽略。因此,在隨后的動力學反應實驗中,選擇酶大小為0.5 mm。研究表明,如采用合適的酶粒徑和酶載荷,可以忽略多孔載體固定化酶的內(nèi)傳質(zhì)限制[21]。
3.2外擴散的影響
在固液酶促反應中,降低外擴散的影響對于提高反應速率具有重要意義,因此,酶促反應需要在最佳攪拌速度下進行[19]。為研究外擴散對PR產(chǎn)率的影響,在50~250 r/min轉(zhuǎn)速下進行外擴散實驗。當溫度為40 ℃,粒徑為0.5 mm,E0=6 U/mL時,攪拌速度對PR產(chǎn)率的影響如圖4所示。由圖4可以看出,當攪拌速度從50 r/min增加到150 r/min,PR產(chǎn)率從20.1%增加到46.2%時,攪拌轉(zhuǎn)速對PR產(chǎn)率有較大的影響,反應受外擴散限制。然而當速度從150 r/min增加到250 r/min時,PR產(chǎn)率變化較小,這意味著外擴散影響可以忽略。因此,本實驗的最佳反應速度是150 r/min。
上述實驗結(jié)果表明,當酶粒徑和攪拌轉(zhuǎn)速分別為0.5 mm和150 r/min時,內(nèi)、外擴散阻力均可以忽略,故所研究的是固有動力學模型。
3.3動力學模型
優(yōu)化式(4)~式(13),優(yōu)化后的動力學模型參數(shù)如表1所示。PR和PS產(chǎn)率模型計算值與實驗數(shù)據(jù)比較如圖5所示,由圖5可以看出,PR和PS的產(chǎn)率分別為97.3%和1.82%,ee%值為96.5%,取得很好的動態(tài)共價動力學拆分效果。由于脂肪酶的高選擇性,PR的產(chǎn)率遠高于PS,從而得到高ee%。影響對映選擇性的另一個因素是底物結(jié)構(gòu)。在本研究中,選擇結(jié)構(gòu)比乙二醇醛二聚體大的2-(苯基甲氧基)乙醛與1,4-二硫-2,5-二醇反應,能更適合于酶的活性位點[6]。
A和B剩余率模擬值和實驗值如圖6所示。由圖6可以看出,由于A、B采用等反應量進料,兩條剩余率曲線重合,A的最終剩余率為0.4%,相應轉(zhuǎn)化率為99.6%。
當條件溫度為40 ℃,粒徑為0.5 mm,攪拌速度為150 r/min,E0=6 U/mL時,P1和B的模擬值和實驗值的比較如圖7所示。由圖7可以看出,模擬值和實驗數(shù)據(jù)之間的平均和最大相對偏差分別為8.9%和65.7%。最大相對偏差出現(xiàn)在低PR產(chǎn)率或A、B剩余率處,例如,在PR的低產(chǎn)率下,即使較小的偏差值也會產(chǎn)生較大的相對偏差,因為較小的PR是分母。盡管最大相對偏差為65.7%,但這并不意味著所建立的反應動力學模型與實驗數(shù)據(jù)擬合得不好,而且模擬值與實驗數(shù)據(jù)之間的偏差分布為零軸對稱,進一步表明,該模型很好擬合了實驗數(shù)據(jù)。酶反應動力學模型對于研究酶反應機制十分重要[21],模型很好擬合了實驗數(shù)據(jù),說明設定的反應機制是正確的。反應式(1)遵循冪定律,反應式(2)和式(3)的酶催化反應,則是帶有產(chǎn)物抑制順序機制。在許多由酶催化的動力學模型中,經(jīng)常使用順序機制[15]。K13y(8)A0為反應式(2)和式(3)的產(chǎn)物P1抑制項,而K13是P1的產(chǎn)物抑制系數(shù)。與其他常數(shù)相比,K13數(shù)值較大,因此P1的產(chǎn)物抑制對反應具有較強的影響(見表1)。
實際上,在建立化學酶動力學模型時,還考慮了底物抑制和非競爭性抑制等因素。不同動力學模型擬合實驗數(shù)據(jù)如表2所示。表2中,模型方程式分母不同,但分子相同。根據(jù)模擬和實驗數(shù)據(jù)之間的平均相對偏差,判斷模型是否合適的標準分為四個等級,即非常好(<10%)、好(<15%)、較差(<20%)和差(>20%)。由表2可以看出,不同模型的差別僅出現(xiàn)在式(6)的分母上,如果添加底物抑制或非競爭性抑制,則式(6)分母將改變。其中,只有模型原型(式(6))可以很好地擬合實驗數(shù)據(jù),而其他5個模型擬合較差。說明P1苯酚產(chǎn)物抑制的假設可以接受,而其他假設,即A和B的非競爭性抑制、PR和PS產(chǎn)物抑制以及CR,CS和D底物抑制的假設是錯誤的。而具有底物抑制作用的脂肪酶介導的ε-己內(nèi)酯合成動力學模型[20],其脲素過氧化氫酶和乙酸都具有較強的底物抑制作用。
對于化學-酶反應,存在三種可能的情況,酶促反應抑制;酶與化學反應抑制;化學反應抑制。前兩種情況增加酶濃度,可減輕由酶促反應引起的抑制,并提高酶促反應的速率。當E0為6 U/mL時,PR的產(chǎn)率為97.3%,而當E0為2 U/mL時,PR的產(chǎn)率僅為89.2%,這表明隨著E0的增加,反應產(chǎn)率增加,即增加脂肪酶的量能加快酶促內(nèi)酯化,使PR產(chǎn)率增加。中間體CR與反應式(1)和式(2)有關(guān),其中反應式(1)生成CR,而反應式(2)消耗CR。因此,在動態(tài)共價動力學拆分系統(tǒng)中,CR的量由兩個反應確定,CR和CS的比較如圖8所示。
由圖8可以看出,在反應的初始階段,CR迅速增加,因為反應式(1)立即開始生成CR,而消耗CR的反應式(2)有延遲。隨著反應的進行,CR升高到一定水平時,反應式(2)的速率迅速增加,并超過反應式(1)的速率。結(jié)果消耗的CR多于生成的CR,CR隨著時間下降。CS與CR相似,但反應式(3)比反應式(2)慢,消耗的CS更少。CS主要通過反應式(1)的可逆反應返回起始原料,因此CS在系統(tǒng)中高于CR。
4結(jié)束語
本研究將可逆半硫代乙縮醛轉(zhuǎn)化與脂肪酶催化對映選擇性內(nèi)酯化反應結(jié)合,構(gòu)成了動態(tài)動力學拆分體系,采用一鍋法絲孢酵母脂肪酶催化,制備了對映純PR。研究結(jié)果表明,其轉(zhuǎn)化率達到99.6%,產(chǎn)率達97.3%,對應體過量百分率達96.5%。本文首次建立的化學-脂肪酶動態(tài)動力學拆分體系的固有動力學模型,很好地擬合了實驗數(shù)據(jù)。反應動力學揭示了酶催化內(nèi)酯化是反應限制環(huán)節(jié),因此增加脂肪酶的量,可以加速反應,從而使PR產(chǎn)率顯著提高。
參考文獻:
[1]Ren Y, Hu L, Ramstrm O. Multienzymatic cascade synthesis of an enantiopure (2R, 5R)-1, 3-oxathiolane anti-HIV agent precursor[J]. Molecular Catalysis, 2019, 468: 52-56.
[2]Zhang Y, Schaufelberger F, Sakulsombat M, et al. Asymmetric synthesis of 1, 3-oxathiolan-5-one derivatives through dynamic covalent kinetic resolution[J]. Cheminform, 2014, 45(24): 3826-3831.
[3]彭燕鴻, 彭燕鴻, 蘇愛秋, 等. 微生物嗜熱脂肪酶研究進展[J/OL]. 食品與發(fā)酵工業(yè): 1-8[2020-10-22]. https: ∥doi. org/10. 13995/j. cnki. 11-1802/ts. 025090.
[4]張燦, 姜國芳, 楊江楠, 等. 多孔材料固定化脂肪酶的研究進展[J]. 分子催化, 2020, 34(4): 378-396.
[5]Chen Y, Zhang X, Zheng G, et al. Preparation of the enantiomerically enriched precursor of lamivudine (3TCTM) via asymmetric catalysis mediated by Klebsiellaoxytoca[J]. Process Biochemistry, 2019, 81(7): 77-84.
[6]Hu L, Schaufelberger F, Zhang Y, et al. Efficient asymmetric synthesis of lamivudine via enzymatic dynamic kinetic resolution. [J]. Chemical Communications, 2013, 49(88): 10376-10378.
[7]Sakulsombat M, Zhang Y, Ramstrm O. Dynamic asymmetric hemithioacetal transformation by lipase-catalyzed γ-lactonization: in situ tandem formation of 1, 3-oxathiolan-5-one derivatives[J]. Chemistry-A European Journal, 2012, 18(20): 6129-6132.
[8]Zhang Y, Hu L, Ramstrom O. Double parallel dynamic resolution through lipase-catalyzed asymmetric transformation[J]. Chemical Communications, 2013, 49(18): 1805-1807.
[9]Pornrapee V, Marcus A, Rikard L, et al. Dynamic combinatorial resolution: direct asymmetric lipase-mediated screening of a dynamic nitroaldol library[J].? Angewandte Chemie International Edition, 2007, 119(6): 9666-968.
[10]李凱. 基于新材料和新策略的洋蔥伯克霍爾德菌脂肪酶固定化及其應用研究[D]. 武漢: 華中科技大學, 2019.
[11]Zhang Y Y, Liu J H. Kinetic study of enantioselective hydrolysis of (R, S)-ketoprofen ethyl ester using immobilized T. laibacchii lipase[J]. Biochemical Engineering Journal, 2011, 54(1): 40-46.
[12]Zhang Y Y, Lu P, Sun Q H, et al. Lipase-mediated direct in situ ring-opening polymerization of ε-caprolactone formed by a chemo-enzymatic method[J]. Journal of Biotechnology, 2018, 281: 74-80.
[13]Bornadel A, Cecilia Orellana kerman, Adlercreutz P, et al. Kinetic modeling of lipase-catalyzed esterification reaction between oleic acid and trimethylolpropane: A simplified model for multi-substrate multi-product ping-pong mechanisms[J]. Biotechnology Progress, 2013, 29(6): 1422-1429.
[14]Mathpati A C, Badgujar K C, Bhanage B M. Kinetic modeling and docking study of immobilized lipase catalyzed synthesis of furfuryl acetate[J]. Enzyme & Microbial Technology, 2016, 84: 1-10.
[15]Yadav G D, Trivedi A H. Kinetic modeling of immobilized-lipase catalyzed transesterification of n-octanol with vinyl acetate in non-aqueous media[J]. Enzyme & Microbial Technology, 2003, 32(7): 783-789.
[16]Zhang Y, Liu J. Purification and in situ immobilization of lipase from of a mutant of Trichosporonlaibacchii using aqueous two-phase systems[J]. Journal of Chromatography B, 2010, 878(11/12): 909-912.
[17]Ulrika T, Martin H, Karin S, et al. Structural, functional and chemical changes in Pseudozymaantarctica lipase B on exposure to hydrogen peroxide[J]. Biochimie, 2010, 92(12): 1867-1875.
[18]Trnvall U, Fürst C M, Hatti-Kaul R, et al. Mass spectrometric analysis of peptides from an immobilized lipase: focus on oxidative modifications[J]. Rapid Communications in Mass Spectrometry, 2010, 23(18): 2959-2964.
[19]Waghmare G V, Chatterji A, Rathod V K. Kinetics of enzymatic synthesis of cinnamyl butyrate by immobilized lipase[J]. Applied Biochemistry and Biotechnology, 2017, 183(3): 792-806.
[20]Zhang Y, Zhao Y, Jiang W, et al. Lipase-catalyzed oxidation of cyclohexanone to form ε-caprolactone and kinetic modeling[J]. ACS Sustainable Chemistry & Engineering, 2019, 7(15): 13294-13306.
[21]Kamble M P, Shinde S D, Yadav G D. Kinetic resolution of (R, S)-α-tetralol catalyzed by crosslinked Candida antarctica lipase B enzyme supported on mesocellular foam: A nanoscale enzyme reactor approach[J]. Journal of Molecular Catalysis B: Enzymatic, 2016, 132: 61-66.
Abstract:?? In order to effectively improve the yield of enantiomeric 1, 3-oxathiolane (PR) of pharmaceutical intermediates, dynamic covalent kinetic resolution was performed in this study to prepare PR and establish its kinetic model. Using one-pot process, enantiopure ((R)-5-acetoxy-1, 3-oxathiolan-2-yl) ethyl benzoate (PR) was synthesized with 99.6% conversion, 97.3% yield and 96.5% ee., and it was performed using an enzyme called Trichosporonlaibachii CBS 5791, with substrates of 2- (phenylmethoxy) acetaldehyde (A), 1, 4-dithiophene-2, 5-diol (B), and phenyl acetate (D). A kinetic model for the resolution was developed for the first time, which fits the experimental data very well. The transformation may follow a power law, and the enzymatic lactonization may follow a sequential mechanism with product inhibition. PR can be synthesized efficiently by the one-pot method, and the mechanism of the reaction is revealed by the kinetic model established.
Key words: kinetic model; lipase; hemithioacetal conversion; 1, 3-oxathiolane; Enzymatic synthesis
猜你喜歡
童話王國·奇妙邏輯推理(2024年5期)2024-06-19 16:03:38
小獼猴智力畫刊(2022年9期)2022-11-04 02:31:42
中學生數(shù)理化·中考版(2022年11期)2022-02-16 07:01:20
中學生數(shù)理化·七年級數(shù)學人教版(2020年10期)2020-11-26 08:24:50
數(shù)學物理學報(2020年2期)2020-06-02 11:29:24
小哥白尼(趣味科學)(2019年6期)2019-10-10 01:01:50
光學精密工程(2016年6期)2016-11-07 09:07:19
發(fā)明與創(chuàng)新(2016年38期)2016-08-22 03:02:52
太空探索(2016年5期)2016-07-12 15:17:55
核科學與工程(2015年4期)2015-09-26 11:59:03
