硅酸鈷鋰正極材料的研究現狀與前景
2021-04-02 00:48:54武金磊馬運昌魏福祥
電源技術 2021年3期
武金磊,馬運昌,魏福祥,張 翼
(中北大學,山西太原038507)
由于能源和環境問題日益突出,基于電動汽車和電網儲能的關鍵技術引起了世界各國的關注,迫切需要開發出具有高能量密度和大功率的新一代鋰電池。電動汽車和大型儲能裝置的工作電壓是由多個單節鋰電池通過串聯和并聯的方式連接提供的。目前市場上主流的單節鋰電池型號為18650 或21700 型,并且單個鋰電池的電壓約為3.7 V。因此,開發具有高能量密度的鋰電池的關鍵是開發具有高電壓的正極材料,因為只有單個鋰電池的工作電壓提高,才可以減少大型儲能裝置中鋰電池的消耗,并有效地提高能量密度。
聚氧陰離子橄欖石LiFePO4材料已經成功商業化,具有很高的安全性和庫侖效率(實際充放電比容量已接近其理論比容量[1]170 mAh/g),但由于其固有的結構缺陷(每個配方單元只能提供一個Li+插入和脫嵌[2]),限制了其應用。傳統的正極材料不能滿足高能量密度鋰離子電池的市場需求。在這種情況下,同為聚陰離子系的硅酸鹽正極材料引起了研究者的很大興趣,每單元正硅酸鹽Li2MSiO4(M = Fe,Co,Mn ...)可以允許兩個Li+插入和脫嵌,并且其Si-O 共價鍵比P-O 共價鍵更強,因此在電解液中具有較高的化學穩定性[3]。在研究過程中,發現硅酸鹽正極材料具有以下缺點:(1)導電性差,小于10-14S/cm,比LiFePO4低3~5 個數量級[4]。(2)Li2FeSiO4在充放電過程中,第二次循環的充電曲線相對于首次循環會顯示出明顯的電壓降。根據充電前后的X 射線衍射(XRD)分析,發現Li2FeSiO4在第一次充電過程中的XRD 特征峰發生偏移并逐漸穩定,表明該材料在充電過程中發生了相變,從結構無序轉變為有序,形成更穩定的結構,且Li2MnSiO4材料在充放電過程中,結構會從有序變為無序,無法恢復。(3)可逆容量低,充放電過程中伴隨著氧陰離子的氧化還原(O2-/On-),很難實現兩個鋰離子的插入和脫嵌。與其他正極材料相比,Li2CoSiO4(LCSO)具有電壓平臺高(高于4 V)、理論比容量高(為325 mAh/g)、電壓平臺循環穩定和熱穩定性良好等優點,有望成為新一代鋰離子電池正極材料。反螢石結構的Li6CoO4材料具有四面體對稱結構,以過渡金屬氧四面體TO4為結構單元,每個化學式有六個鋰位,比硅酸鹽具有更高的鋰位與變價活性元素摩爾比,從而在理論上具有成倍提高比容量的可能性。以四面體MO4基元為出發點探索新型高鋰氧化物正極材料的設計不僅具有學術上的研究意義,同時也將為解決鋰離子電池的產業瓶頸奠定科學基礎。其首次充電比容量可達到600 mAh/g 以上,但它們與硅酸鹽正極材料面臨著相似的問題,具有電導率低(小于10-14S/cm)、電壓平臺低、可逆容量低(首次放電比容量只有27 mAh/g)和結構不穩定等缺點。因此,對于LCSO 正極材料的研究對今后開發出具有更高容量的高鋰材料Li6CoO4也具有重要意義。
本文聚焦LCSO 的多晶型結構,通過不同的合成方法和手段改善其物理和電化學性能,總結其近年來的研究進展。
1 Li2CoSiO4的合成
1.1 合成方法
(1)固相反應。傳統的固相反應是合成Li2MSiO4/C 最常用的方法,該方法具有成本低、效率高、可延展性好等優點。在固相反應過程中,將反應物充分混合、研磨得到前驅體,然后加熱得到Li2MSiO4/C 樣品。常用的起始合成材料是含有鋰、鐵、錳、鈷和硅酸鹽元素的化合物,例如氧化物、水合物、硅酸鹽/硫酸鹽/氯化物、草酸鹽、乙酸酯、碳酸鹽等。有時也使用正硅酸四乙酯[TEOS、Si(OC2H5)4]和Li2SiO3。可添加炭黑、蔗糖、葡萄糖、抗壞血酸、煤、瀝青[5]、碳納米球(CNS)、碳納米管(CNT)[6]、還原氧化石墨烯(RGO)、氧化石墨烯(GO)、乙炔黑[7]等碳源,以提高正極材料的導電性。采用這種技術,Li2MSiO4通常在600~900 ℃高溫下煅燒8~12 h 下合成,但除研磨和退火工藝外,固相反應存在組分混合不均勻、形貌不規則、顆粒生長不受控制、團聚嚴重、加熱時間長等缺點。
(2)溶膠-凝膠法。溶膠-凝膠法以無機材料或金屬醇鹽為前驅體,在液相中均勻混合,進行水解和縮合化學反應。檸檬酸、蔗糖、聚乙二醇、聚乙烯醇、酒石酸和抗壞血酸等螯合/絡合劑常用于溶膠-凝膠技術。合成溫度通常為600~800 ℃,反應時間為5~15 h,通過溶膠-凝膠技術可以得到不同形貌的Li2MSiO4,如球形、介孔、團聚體、蜂窩狀等。但是用該方法合成的凝膠結構在熱處理過程中易崩塌,其原因是加熱速率過快和Li2MSiO4結晶度低[8],導致其在高倍率充放電下循環穩定性差。為了克服這些問題,水熱合成受到了研究者的高度重視。
(3)水熱反應。目前制備Li2CoSiO4的主要方法是水熱法合成。水熱反應具有簡單、清潔、成本低、能耗低等優點,適用于在密閉條件下制備高質量的多晶材料系統[9]。一般來說,水熱過程在150~200 ℃的溶劑中進行24~96 h。Gong等[10]首次采用水熱反應合成Li2CoSiO4,并以去離子水為反應介質溶劑,獲得的Li2CoSiO4由約2 μm 的單個顆粒組成,初始放電比容量為75 mAh/g。Lyness 等[11]提出了以去離子水和乙二醇為溶劑的水熱合成鋰硅源的新方法,同時對合成材料的結構給出了明確定義,所獲得的材料被定義為βⅠ-Li2CoSiO4(Pbn21)可提供60 mAh/g 的初始放電比容量。He 等[12]報道了以碳/二氧化硅(MCS)為模板制備介孔Li2CoSiO4復合材料,但是它的放電比容量只有33 mAh/g。
張志峰等[13]總結了不同的水熱方法并進行了改進。通過改進的水熱反應合成純度較高的βII-Li2CoSiO4多晶型材料,使用LiOH·H2O、SiO2和CoCl2·6 H2O(4∶1∶1)作為合成初始材料。在該合成中,將去離子水用作鋰和硅源的溶劑,將乙二醇用作鈷源的溶劑,去離子水與乙二醇的比例為2∶1,然后將兩種溶液混合并攪拌以形成Li2CoSiO4前驅體,再將前驅體轉移至高壓反應釜中,在150 ℃下加熱72 h,得到的材料由50 nm 左右的單個顆粒組成。與傳統的水熱法相比,該技術合成的材料具有較小的粒徑且分布均勻。杜文強等[14]在張志峰水熱反應的基礎上,通過借鑒Xu 等人對LiMn0.6Fe0.4PO4的研究[15],發現通過改變水熱反應中LiOH 和SiO2的濃度以及乙二醇(EG)與去離子水的比例,能夠合成出具有不同形貌的LCSO 材料。合成路徑如圖1 所示。通過改變去離子水與乙二醇的比例可以改變水熱反應過程中混合物的粘度。在較高的粘度下,納米顆粒傾向于長成次級顆粒的大聚集體,低粘度促進分散的初級納米顆粒易于形成小尺寸的納米結構,因此通過改變反應物的黏度可以合成出具有不同形態的LCSO顆粒。通過控制前驅體溶液的[OH-]濃度和黏度,成功合成出了點狀、條狀和片狀的LCSO/C 納米顆粒。

圖1 兩步法合成Li2CoSiO4/C 納米復合材料示意圖
(4)其他合成方法。微波熱合成法[16]可以讓目標材料在短時間內直接吸收電磁/微波能量(自加熱),這種方法與其他方法相比耗能較低,具有簡便且高效的優點,可以在600~700 °C 下反應6~20 h 制備出納米團聚體或球體材料,在室溫以上(60°C)可獲得具有高離子傳導性的正極材料。彭忠東等在Li2FeSiO4合成中引入微波外場的作用,通過高能球磨結合微波加熱制備Li2FeSiO4/C 材料。其他合成方法還包括原位模板法[17]、噴霧熱解法、超臨界流體法[7]等。
1.2 碳包覆
導電碳涂層是提高Li2CoSiO4電子導電性的有效而簡單的策略。此外,碳包覆還可以防止顆粒接觸并抑制顆粒聚集。通常,碳包覆方法有兩種。一種是原位碳包覆:混合前驅體和碳源,然后在高溫下煅燒,該方法可防止顆粒團聚,獲得具有均勻碳涂層的復合材料。另一種是非原位碳包覆:首先合成不含碳的原料,然后與碳混合。原料與碳源通過球磨/研磨混合。如果碳源不是炭黑,則需要進行熱處理。目前常見的碳源也分為兩類,一類是有機碳源,如檸檬酸、纖維素、蔗糖和聚乙二醇(PEG),另一種是無機碳源,例如炭黑。
早期,研究者使用炭黑和石墨作為Li2CoSiO4正極材料的碳源,并通過球磨方式將其混合。然而,僅通過球磨方式混和碳源和Li2CoSiO4前驅體獲得的Li2CoSiO4/C 復合材料的電化學性能并沒有明顯改善。之后人們陸續嘗試了幾種不同的碳涂覆方法,包括在2013 年使用有序的介孔碳/二氧化硅(MCS)框架作為硅酸鹽前體和碳源以及在2017 年使用多壁碳納米管[18],以改善Li2CoSiO4的電化學性能,但都效果不大。
直到2018 年,張志峰等人以蔗糖為碳源,通過球磨的方式將蔗糖與Li2CoSiO4材料混合,Li2CoSiO4通過水熱反應制備,將Li2CoSiO4與蔗糖的混合物送入管式爐在600 ℃下煅燒1 h。合成方法如圖1 所示,通過這種碳包覆方法獲得的Li2CoSiO4/C 復合材料的電化學性能得到較大提升,首次充放電比容量分別為226 和112 mAh/g,而未進行碳包覆的Li2CoSiO4材料首次充放電比容量僅為82.4 和30.2 mAh/g,電壓平臺介于2.5~4.6 V[13]。其電化學性能提升的原因在于在高溫下由蔗糖碳化形成的碳涂層,不僅在活性顆粒之間提供了導電連接,從而促進了電子和離子的遷移,而且在熱處理過程中減小了活性材料的粒徑。它可以縮短鋰離子的擴散路徑,促進相鄰粒子之間的良好接觸并減少極化。
1.3 摻雜
包覆只能從外部提高硅酸鹽材料的導電性,而離子摻雜則是從根本上改變鋰離子的內部結構,從而提高鋰離子在材料中的擴散效率。從結構穩定性、可逆性、倍率性能和循環等方面研究發現,離子摻雜是提升其電化學性能的有效途徑。因此,為了提高Li2CoSiO4的性能,Co 位、Li 位、Si 位和O位等各種摻雜技術在Li2CoSiO4的摻雜中得到了廣泛的應用。
硅酸鹽正極材料發展緩慢的主要原因是電子和離子導電性差。一些研究人員已經計算出鈉化合物具有更高的電子和離子導電性,而鋰化合物具有更高的脫鋰電壓和更好的循環穩定性。因此,為了提高LCSO 的電化學性能,可以在Li位進行適當的Na 摻雜。
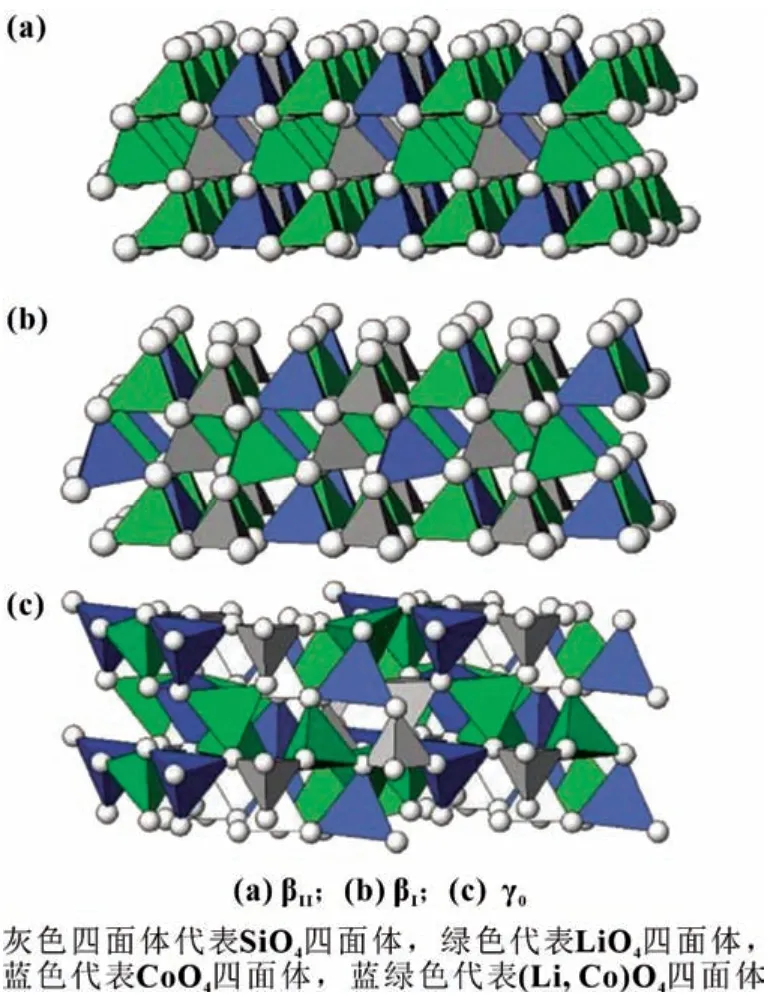
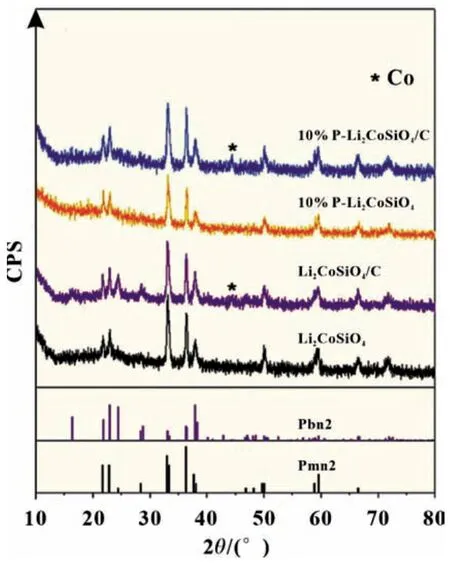
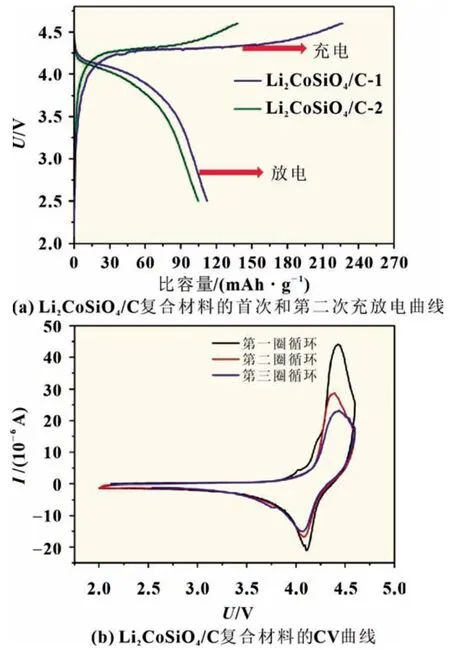
在Co 位進行適當的離子摻雜可以改變Li-O-Co 鍵的強度,引起材料晶胞體積的變化,但不會改變LCSO 的晶體結構。此外,Co 位摻雜可以降低電化學極化,有利于Li+的插入和提取。Co 位摻雜常用的元素有Fe、Mn 和Mg 等。這個類型可表示為Li2Co1-xMxSiO4(M=Fe,Mn,Mg)(0 Si位摻雜可以抑制SiO44-的強絕緣性,提高LCSO 的電導率,從而提升其電化學性能。張等以磷酸為磷源,通過水熱法成功合成了Li2CoSi0.9P0.1O4/C 復合材料[13],其初始充電比容量為270 mAh/g,放電比容量為144 mAh/g。在進一步了解聚陰離子強絕緣性的基礎上,杜等[21]通過系統地改變Al 的摻雜量,研究了碳包覆和Al 摻雜對LCSO 電化學性能的影響,發現Li2CoSi0.96Al0.04O4具有最佳的電化學性能,其首次充放電比容量分別為331 和140 mAh/g。表1 對近年來文獻中Li2Co-SiO4材料的性能做了匯總。 表1 文獻報道的Li2CoSiO4材料第一次充放電比容量 Li2MSiO4化合物具有與Li3PO4相似的結構,都屬于四面體構型。它由正交的O2-離子(扭曲的六邊形緊密堆積形式)組成,其中陽離子占據四面體空隙的一半。陰離子通過與O2-共頂角連接。這種以四面體氧化物為結構單元的化合物具有豐富的晶體結構。當前存在五種已知類型,可以將其大致分為β 和γ,分別對應于Li3PO4的β 相和γ 相。在β 型結構中,所有四面體指向相同并垂直于O2-的緊密堆積面,并且四面體通過共同的頂部相連;在γ 型結構中,每個四面體都作為結構單元堆疊在一起,中間的四面體的方向與另外兩個相反,它們以共線方式連接。根據陽離子在四面體中排列順序的不同,會有不同的結構和晶型,進而將β 和γ 結構分為βⅠ、βⅡ、γ0、γⅡ和γs。 目前我們知道LCSO 存在多種形態,包括βI、βII和γ0三種不同溫度下合成的形態[22],三個相的結構如圖2 所示。通過水熱法合成得到βII相,然后把βII相在700 ℃高溫處理2 h 得到βI相。將βII相1 100 ℃處理2 h,然后降至850 ℃,最后淬火至室溫得到γ0相[11]。它們在高溫時很容易發生相轉換,例如在600 ℃碳包覆時會出現βII相向βI相的轉換,導致很難得到βII純相。LCSO 化合物屬于四面體結構。一般在材料的晶體結構中,氧原子呈現出輕微扭曲的密排六邊形結構。Li、Co、Si 和氧原子分別形成四面體,其中一半四面體中心被陽離子占據,從而避免四面體之間的面共享。Armstrong 等通過rietveld 精修LCSO 三種不同結構的X 射線和中子粉末衍射圖[22],發現βII相中存在明顯的陽離子占位無序。在βII相的理想構型中,Co 和Li 分別分布在四面體的2 a 和4 b 處。而實際合成的βII相過渡金屬2 a 位置完全被Li 占據,而4 b 的位置基本上被Co 和Li 等同占據,βII相中的Li/Co 混排程度比較劇烈,βI相中的Li/Co 混排程度相對較弱,而γ0相中沒有Li/Co 混排。之后通過Li-MAS-NMR 譜(核磁共振譜)進一步證實了這一結果。 圖2 Li2CoSiO4 三種不同相的晶體結構 Noriaki 等[23]通過900 ℃高溫固相反應合成了具有Pbn21(βI)相的LCSO。從DTA 曲線發現,LCSO 在540 ℃開始由βII相轉變為βI相。同時,通過高溫原位XRD 證實了Pbn21到Pmn21的可逆二階相變,解釋為Co 和Li 原子的有序無序轉變。Li/Co 混排引起Pbn21相有序到無序的轉變。Co 亞晶格的無序是由于與Li 原子的位置交換造成的,這與水熱反應合成材料的報道一致。過渡金屬的有序度直接影響到LCSO 不同相的結構穩定性、高壓性能和熱處理工藝。Wu 等[24]通過第一原理計算研究了LixCoSiO4(0≤x≤2)的結構和電子性能。結合不同脫鋰狀態下的形變電荷密度和電子態密度,發現Co-O鍵的鍵長隨著鋰離子的脫嵌和鈷離子的氧化顯著減小,而Si-O 鍵的鍵長基本沒有變化。這表明在鋰化-脫鋰過程中,硅與氧原子的成鍵性能沒有發生變化;同時證明了Li2CoSiO4的結構穩定性實際上是強Si-O 共價鍵作用的結果。根據Zhang等[25]的計算結果,Co 基硅酸鹽的晶格框架不同于鐵基和錳基體系的二維層狀結構,是一種三維框架結構。脫鋰過程中產生的Peierls 畸變可能是制約鈷基材料充放電能力提高的關鍵微觀因素。 在第一性原理密度泛函理論(DFT)計算的基礎上,研究不同離子摻雜對LCSO 結構的影響,用于指導實驗合成。所有結構基于DFT 理論的VASP 軟件計算,并采用PAW 贗勢,交換關聯函數GGA 和PBE 方法。 通過構建P 在LCSO 的Si 位摻雜模型進行研究,LCSO 中P 取代Si 的原子模型用2×1×1 的上層結構構成。該結構包含8 個分子式,其中一個被P 取代。由于P 比Si 少一個電子,因此計算了帶負電態的P 摻雜體系。然后,對相關模型進行了第一原理結構優化。通過比較Li1/2CoSiO4和Li1/2CoSi7/8P1/8O4在75%脫鋰相中的模型以及CoO4四面體在兩種結構中的鍵長分布可以看出,在未摻雜模型中,Li+具有對稱的階梯分布,四面體形成有序的電荷層,這表明它具有一個帶有Peierls 畸變的高應力網絡,在P 摻雜模型中,P 摻雜位點可以形成排斥性正電荷中心,從而可以抑制脫鋰相Peierls 畸變的非對稱應力。同時,通過比較P 摻雜于未摻雜計算模型的電子結構計算,發現P 摻雜可以減小LCSO 的能帶間隙[13]。 通過濕式球磨混合LCSO 前驅體和蔗糖并在氬氣中于600 ℃煅燒1 h,制備0.1P-LCSO/C,0.04Al-LCSO/C 復合材料,它們表現出主要的正交相(Pmn21-DP)。其XRD 特性如圖3 所示(LCSO 在20°~25°之間的XRD 譜中有兩個特征峰,在本工作中標記為DP 相,在20°~25°之間的XRD 譜中有三個特征峰被標記為TP 相)。從之前的工作中我們了解到,LCSO 在540 ℃開始從DP 相轉變為TP 相,即在未摻雜的情況下,LCSO 在高溫碳包覆后表現為DP 和TP 的混合多晶型相。但是我們發現,在LCSO 的Si 位進行P、Al 摻雜后,即使經過600 ℃高溫碳包覆后,得到的產物DP 相仍然占主導地位。這表明在Si位進行離子摻雜可以有效地抑制TP 相的產生。以前我們認為Si 位摻雜的主要作用是改變LCSO 的電子傳輸特性,然而,通過實驗發現Si 位摻雜不僅可以改變四面體多晶型的熱穩定性,而且有助于獲得LCSO 純相材料。此外,摻雜[13]過程也給我們帶來了一些困惑。與純相材料相比,摻雜后形成的復合材料在碳包覆過程中更容易產生Co 或Co 相關雜質。通過分析,推測TP 相的結構穩定性優于DP 相,而在Si 位進行摻雜后會抑制TP 相的生成,因而導致在Si位進行摻雜后的復合材料相對于未摻雜的LCSO 在碳包覆過程中更易于還原Co。 圖3 LCSO、LCSO/C、0.1P-LCSO和0.1P-LCSO/C 的XRD 圖譜 圖4 Li2CoSiO4/C 復合材料的充放電曲線和CV曲線 在2.5~4.6 V 電壓平臺內,取電流密度5 mA/g 對LCSO/C復合材料進行充放電測試。Li2CoSiO4/C 復合材料的第一次充電比容量為226 mAh/g,放電比容量為112 mAh/g,具有約4.25 V 的充電平臺和約4.1 V 的放電平臺,如圖4(a)所示。在第二圈循環中,電壓平臺沒有發現明顯的變化。與已有的文獻報告相比,在2.5~4.6 V 的工作電壓內,0.7Li+的可逆脫嵌遠高于文獻報道的75 mAh/g(0.46Li+)LCSO 的最大容量。 通過對LCSO/C 復合材料的循環伏安(CV)曲線進行分析[26],如圖4(b)所示,發現在第一圈循環中LCSO/C 的氧化峰在4.3 V 左右,還原峰在4.15 V 左右;4.3 V 的氧化峰與4.15 V的還原峰代表了LCSO 材料中Co2+離子的氧化和還原,在接下來的兩次循環中也存在同樣位置的氧化還原峰。而氧化峰和還原峰的強度代表著氧化還原反應的難易程度,峰的強度越大,氧化還原反應越容易。氧化峰和還原峰的強度比值越接近于1,代表電池可逆性越好,放電容量越接近充電容量。從CV 曲線中,我們發現LCSO/C 復合材料的氧化峰比還原峰強得多,代表這個材料的可逆性不是很好,即放電容量遠小于充電容量,這個結論可以通過充放電曲線得到證實。同時還發現,在第二圈循環過程中的氧化峰與第一圈循環相比強度迅速下降,這表明材料的循環穩定性較差。體現在在充放電曲線中為第二圈的充電容量遠小于第一圈。 對比摻雜P 后材料的充放電曲線和CV 曲線,如圖5 所示[26],發現其存在和未摻雜材料類似的情況,即在充放電過程中充電容量遠大于放電容量,表現在CV 曲線中為氧化峰強度遠大于還原峰,且在隨后的循環過程中,氧化峰和還原峰都有不同程度的下降,這應該就是LCSO 材料循環穩定性差的原因。0.1P-LCSO/C 材料的充放電比容量[13]分別為270 和143 mAh/g,基本上能夠實現一個鋰離子的插入和脫嵌。0.04Al-LCSO/C 的首次充放電比容量[21]分別為331 和140 mAh/g。這表明在Si 位進行P、Al 摻雜在一定程度上雖然能夠提升LCSO 材料的電化學性能,但對其循環穩定性改善程度有限。 圖5 P-Li2CoSiO4/C復合材料的充放電曲線和CV 曲線 為了搞清楚對Si 位進行摻雜相對于純相材料電化學性能提升的原因,以及純相材料電化學性能差的原因,通過XRD 測試了充放電前后的0.04Al-LCSO/C 和經過一次循環的LCSO/C 復合材料,如圖6 所示[27]。發現在充放電循環過程中LCSO/C 復合材料的一些XRD 特征峰消失,表明LCSO/C 復合材料的結構在充電過程中發生了有序到無序的轉變。同時,由于LCSO/C 復合材料的結構無序化,導致鋰離子遷移路徑紊亂,難以將鋰離子重新插入到LCSO/C 顆粒中,因而它在首圈循環中具有較大的不可逆容量。而且,與未經過充放電測試的原始LCSO/C 材料相比,發現在第一次充放電循環后,LCSO/C 材料的XRD 特征峰強度變弱,表明其結晶度在充放電循環過程中降低。結合充放電曲線分析,LCSO/C 復合材料在放電過程中不能保持長程有序,導致材料的庫侖效率低,即放電容量遠小于充電容量。另外,由于結晶度連續降低,導致Li2CoSiO4/C 的循環穩定性差。與未摻雜材料相比,摻雜后的材料在充電的末態仍能保持原來的結構,但與未經過充放電測試的材料進行對比,發現它們的XRD 特征峰強度降低,即結晶度比未充電時有所降低。在LCSO 的Si 位進行P 和Al摻雜,與未摻雜材料相比,在一定程度上能夠保持結構的穩定性,降低材料在充放電過程中結晶度下降的趨勢,從而具有相對較高的充放電性能和循環穩定性。 圖6 0.04Al-LCSO/C 和LCSO的XRD 對比圖 LCSO 正極材料電化學性能差的根本原因在于其在充放電過程中,結構上會發生有序到無序的轉變,雖然這個過程是可逆的,但在充放電過程中,隨著循環的進行,其結晶度會不斷下降直至消失,導致其循環穩定性差。目前改善LCSO材料電化學性能最有效的措施是在Si 位進行離子摻雜,一方面可以改善SiO44-的強絕緣性,另一方面,能夠在一定程度上抑制LCSO 在充放電過程中結構有序到無序的相轉變,降低其結晶度下降趨勢。但這仍未從根本上解決LCSO 在長循環過程中結構有序到無序的相轉變,因而只能在一定程度上改善LCSO 的循環穩定性。今后我們的工作重點是,找到更好的摻雜元素來穩定LCSO 材料的結構。之前我們了解到Li2MnSiO4材料在充放電過程中結構會發生有序到無序的相轉變,且不能恢復,但是有研究人員對Li2MnSiO4材料進行Fe摻雜,可以改善其結構穩定性,在充放電過程中保持其結構有序性。這證明,找到合適的摻雜元素,保證LCSO 材料在充放電過程中保持其結構長程有序性是可行的。此外,LCSO材料的低溫相存在很大程度的Li/Co 混排,即Li、Co 占位無序,在充放電過程中會影響鋰離子遷移路徑,進而影響其電化學性能。而通過計算和實驗表明,在高溫下可以獲得占位有序的、具有晶體結構的LCSO 材料,目前尚未發現這方面的研究。這為我們在高溫下合成出占位有序的晶體結構材料,從而改善LCSO 的電化學能提供了一定的指導意義。 隨著鋰離子電池在電動汽車(EVs)和大型電能儲存設備上的大規模應用,迫切需要開發出能量和功率密度更高、循環穩定性更好的新一代鋰離子正極材料。通過改善合成工藝,使用碳包覆和離子摻雜等改性手段對LCSO 材料的電化學性能進行改善,有望使其成為下一代動力鋰離子電池的重要候選材料之一。
2 聚陰離子結構
2.1 Li2CoSiO4的多晶結構及其相變
2.2 摻雜對Li2CoSiO4結構的影響
3 電化學性能


4 結論
猜你喜歡
建材發展導向(2021年14期)2021-08-23 00:56:16
哲學評論(2021年2期)2021-08-22 01:53:34
紡織科技進展(2021年3期)2021-06-09 08:07:14
中華詩詞(2019年7期)2019-11-25 01:43:04
模具制造(2019年3期)2019-06-06 02:10:54
纖維復合材料(2018年3期)2018-04-25 07:22:58
電子測試(2017年11期)2017-12-15 08:57:13
影視與戲劇評論(2016年0期)2016-11-23 05:26:01
現代企業(2015年9期)2015-02-28 18:56:50
應用化工(2014年10期)2014-08-16 13:11:29
