GRIN1突變相關(guān)發(fā)育遲緩患兒臨床特征和基因突變特點(diǎn)分析
2021-03-09 11:49:52盧亞亞姚如恩王依柔張倩文王秀敏
檢驗(yàn)醫(yī)學(xué) 2021年2期
盧亞亞, 丁 宇, 姚如恩, 王依柔,張倩文, 李 群, 王 劍, 王秀敏, 婁 丹
(1.河南科技大學(xué)第一附屬醫(yī)院兒科,河南 洛陽(yáng) 471000;2.上海交通大學(xué)醫(yī)學(xué)院附屬上海兒童醫(yī)學(xué)中心內(nèi)分泌代謝科,上海 200127;3.上海交通大學(xué)醫(yī)學(xué)院附屬上海兒童醫(yī)學(xué)中心分子診斷實(shí)驗(yàn)室,上海 200127)
興奮性氨基酸受體是哺乳動(dòng)物中樞神經(jīng)系統(tǒng)重要的興奮性神經(jīng)遞質(zhì)受體,負(fù)責(zé)調(diào)節(jié)神經(jīng)系統(tǒng)功能。谷氨酸(glutamic acid,Glu)在所有興奮性氨基酸遞質(zhì)中含量最為豐富。Glu受體分為離子型和代謝型。離子型 Glu受體分為N-甲基-D-天冬氨酸(N-methyl-D-aspartate,NMDA)受體和非NMDA受體[1]。NMDA受體是Glu受體的主要亞型,是由不同亞基構(gòu)成的異四聚體,有3種亞基[NR1、NR2(NR2A-2D)、NR3(NR3A、NR3B)][2]。NMDA受體主要參與學(xué)習(xí)記憶、突觸可塑性、興奮性神經(jīng)毒性等生理病理過(guò)程。GRIN1基因編碼NMDA受體的NR1亞基。NR1亞基是基本功能亞單位,如缺失會(huì)嚴(yán)重影響NMDA受體的功能[3]。GRIN1基因變異可引起癲癇性腦病、精神發(fā)育遲緩、語(yǔ)言落后、肌張力減退、運(yùn)動(dòng)障礙、腦萎縮。該基因變異非常罕見(jiàn)。本研究對(duì)2例GRIN1基因新發(fā)突變患兒的臨床表型及分子特征進(jìn)行分析,探討GRIN1基因突變?cè)趦和l(fā)育遲緩中的價(jià)值。
1 材料和方法
1.1 病例介紹
病例1,女,1歲,以“發(fā)育遲緩數(shù)月”為主要癥狀就診。患兒系第2胎第2產(chǎn),足月自然分娩出生,出生時(shí)無(wú)窒息搶救史,出生體質(zhì)量3.0 kg。當(dāng)?shù)蒯t(yī)院的新生兒臨床檢查未發(fā)現(xiàn)異常。4月齡時(shí),因不會(huì)抬頭至當(dāng)?shù)蒯t(yī)院進(jìn)行檢查,體檢發(fā)現(xiàn)患兒無(wú)追視,不會(huì)對(duì)人微笑,雙下肢肌張力低,膝腱反射正常。實(shí)驗(yàn)室檢查結(jié)果:肝、腎功能正常,血氨及乳酸正常,肌酸激酶及其同工酶正常,血常規(guī)、尿常規(guī)、糞便常規(guī)、血糖、血電解質(zhì)、25-羥基維生素D3均無(wú)異常,串聯(lián)質(zhì)譜檢查所有氨基酸和酰基肉堿譜均無(wú)明顯異常。頭顱磁共振成像(magnetic resonance imaging,MRI)檢查正常。擬診斷:發(fā)育落后。當(dāng)?shù)蒯t(yī)院給予綜合康復(fù)及對(duì)癥支持治療,癥狀無(wú)改善。患兒12月齡時(shí)至上海交通大學(xué)醫(yī)學(xué)院附屬上海兒童醫(yī)學(xué)中心內(nèi)分泌醫(yī)學(xué)遺傳聯(lián)合門(mén)診就診。體格檢查:身高76 cm,體質(zhì)量9 kg,頭圍44 cm,無(wú)特殊面容,對(duì)人和物無(wú)反應(yīng),抬頭不穩(wěn),不會(huì)獨(dú)坐,不會(huì)翻身,無(wú)咿呀作語(yǔ),心、肺聽(tīng)診無(wú)異常,右耳聽(tīng)力檢查欠佳,視力正常。頭顱MRI檢查未見(jiàn)異常。染色體核型、肝功能、堿性磷酸酶、葡萄糖、乳酸、電解質(zhì)和尿液分析均正常。血氨、乳酸、血清游離鈣檢測(cè)結(jié)果及血液串聯(lián)質(zhì)譜分析均正常。
病例2,男,7歲4個(gè)月,以“發(fā)現(xiàn)身材矮小數(shù)年并發(fā)育遲緩”為主要癥狀就診。患兒系第1胎第1產(chǎn),足月自然分娩出生,出生時(shí)無(wú)窒息搶救史,出生體質(zhì)量3.3 kg,當(dāng)?shù)蒯t(yī)院新生兒臨床檢查無(wú)異常,逐漸出現(xiàn)發(fā)育落后,伴生長(zhǎng)速率減慢,具體不詳。當(dāng)?shù)蒯t(yī)院實(shí)驗(yàn)室檢查結(jié)果:血常規(guī)、尿常規(guī)、糞常規(guī)、肝功能、腎功能、血氨、乳酸、肌酸激酶及其同工酶、血糖、血電解質(zhì)無(wú)異常,串聯(lián)質(zhì)譜檢查所有氨基酸和酰基肉堿譜均無(wú)明顯異常,頭顱MRI平掃正常。擬診斷:發(fā)育落后、矮小癥。給予對(duì)癥支持治療,癥狀改善緩慢,發(fā)育仍明顯落后于正常同齡兒童。患兒7歲4個(gè)月時(shí)至上海交通大學(xué)醫(yī)學(xué)院附屬上海兒童醫(yī)學(xué)中心內(nèi)分泌醫(yī)學(xué)遺傳聯(lián)合門(mén)診就診。體格檢查:身高116 cm,體質(zhì)量18.4 kg,頭圍49 cm,對(duì)人和物有反應(yīng),行走自如,會(huì)單腳跳,簡(jiǎn)單詞匯,不能連成句,心、肺聽(tīng)診無(wú)異常,四肢活動(dòng)可,視力正常,聽(tīng)力正常。頭顱MRI檢查未發(fā)現(xiàn)異常。染色體核型、肝功能、堿性磷酸酶、葡萄糖、乳酸、電解質(zhì)和尿常規(guī)檢測(cè)均正常。甲狀腺功能正常,胰島素樣生長(zhǎng)因子1(insulinlike growth factor 1,IGF-1)99.4 ng/mL(降低),胰島素樣生長(zhǎng)因子結(jié)合蛋白-3(insulinlike growth factor-binding protein 3,IGFBP-3)3.96 μg/mL(降低),血氨、乳酸、血清游離鈣檢測(cè)結(jié)果均正常。
本研究經(jīng)上海交通大學(xué)附屬上海兒童醫(yī)學(xué)中心倫理委員會(huì)審查批準(zhǔn),患兒監(jiān)護(hù)人均知情同意。
1.2 方法
采集2例患兒及父母外周血,用Lab-Aid DNA分離試劑盒(廈門(mén)致善生物科技股份有限公司)抽取DNA,使用Qubit 2.0 熒光計(jì)(美國(guó)Life Technologies公司)測(cè)定DNA濃度,-20 ℃保存。采用SureSelect靶序列富集試劑盒(美國(guó)Agilent公司)制備接頭連接的文庫(kù),采用包含2 742個(gè)基因的XT Inherited Disease panel(美國(guó)Agilent公司)制備并獲得捕獲文庫(kù),通過(guò)IlluminacBot工作站(美國(guó)Illumina公司)的等溫橋式擴(kuò)增產(chǎn)生DNA簇,最后用HiSeq 2500系統(tǒng)(美國(guó)Illumina公司)進(jìn)行靶向基因測(cè)序(targeted gene sequencing,TGS)。目標(biāo)區(qū)域平均測(cè)序深度為135×,20×以上區(qū)域?yàn)?8.85%。測(cè)序數(shù)據(jù)經(jīng)Sequence Control軟件(美國(guó)Illumina公司)評(píng)估,合格后采用NextGENe軟件對(duì)數(shù)據(jù)進(jìn)行初步處理,然后采用Variant Analysis軟件(美國(guó)Ingenuity Systems公司)進(jìn)行生物信息學(xué)分析。選擇腦發(fā)育異常的臨床癥狀作為篩選候選變異的指標(biāo)。對(duì)篩查出的基因進(jìn)行Sanger測(cè)序驗(yàn)證。
2 結(jié)果
2.1 2例發(fā)育遲緩患兒基因檢測(cè)結(jié)果
病例1:GRIN1基因存在“錯(cuò)義變異c.1672T>G,p.Phe558Val(雜合)”,患兒父母親該位點(diǎn)為正常基因型。該變異為新發(fā)現(xiàn)的變異,經(jīng)Alamut功能軟件(瑞士SOPHiA GENETICS公司)預(yù)測(cè)可能會(huì)影響蛋白結(jié)構(gòu)域的功能。按美國(guó)醫(yī)學(xué)遺傳學(xué)和基因組學(xué)學(xué)會(huì)(American College of Medical Genetics and Genomic,ACMG)變異分類(lèi)標(biāo)準(zhǔn),歸類(lèi)為“可能致病性”變異。見(jiàn)圖1。
病例2:GRIN1基因存在“錯(cuò)義變異c.1852G>A,p.Gly618Ser(雜合)”。患兒父母親該位點(diǎn)為正常基因型。該變異為新發(fā)現(xiàn)的變異,經(jīng)Alamut功能軟件預(yù)測(cè)可能會(huì)影響蛋白結(jié)構(gòu)域的功能。按照ACMG變異分類(lèi)標(biāo)準(zhǔn),歸類(lèi)為“可能致病性”變異。見(jiàn)圖1。
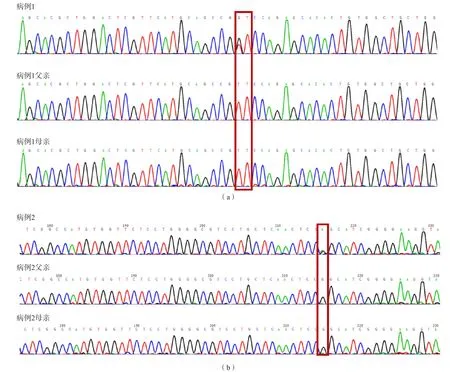
圖1 2例發(fā)育遲緩患兒及其父母GRIN1基因序列
2.2 臨床隨訪
病例1隨訪至2歲,期間排除其他器官、系統(tǒng)畸形,行心臟、腹部超聲等均未見(jiàn)異常。行康復(fù)治療,全身肌張力低下,運(yùn)動(dòng)發(fā)育遲緩,不能獨(dú)坐,可伸手取遠(yuǎn)處物品,有眼神交流,不會(huì)說(shuō)話。身高、體質(zhì)量與同年齡兒童相仿。
病例2隨訪至8歲,身高增長(zhǎng)至119 cm,智力發(fā)育無(wú)明顯改善,明顯落后于正常同齡兒童。
2.3 文獻(xiàn)報(bào)道的GRIN1基因突變類(lèi)型和患兒臨床表現(xiàn)分析
對(duì)本研究的2例及文獻(xiàn)報(bào)道的37例GRIN1基因突變相關(guān)發(fā)育落后患兒的GRIN1基因突變類(lèi)型和臨床表現(xiàn)進(jìn)行分析,具體見(jiàn)圖2及表1。
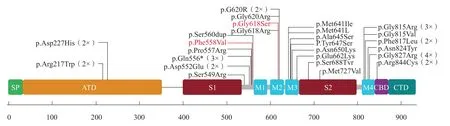
圖2 GRIN1基因突變區(qū)域分布
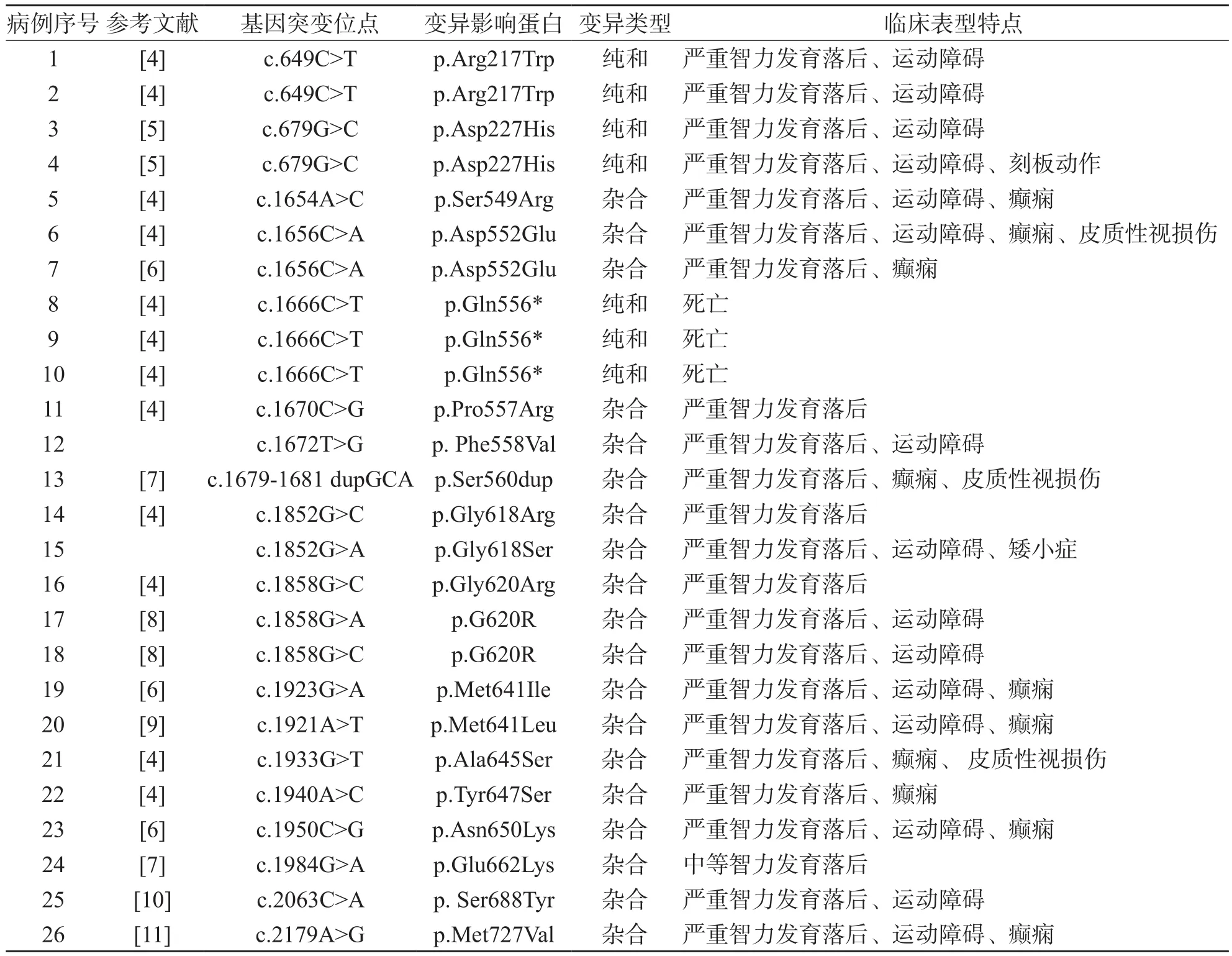
表1 GRIN1基因突變患兒的臨床表型分析
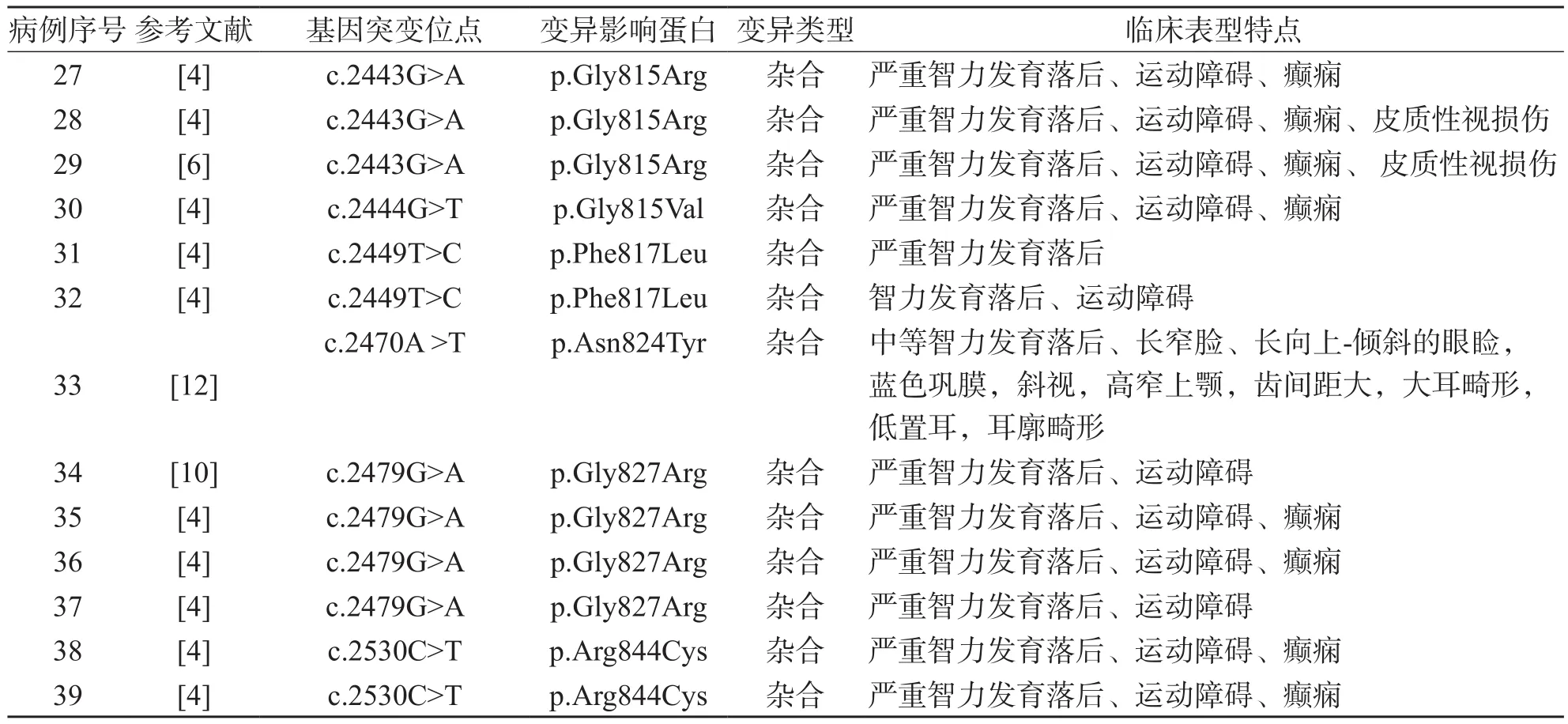
續(xù)表1
3 討論
智力障礙或全面發(fā)育遲緩是一大類(lèi)具有高度臨床和遺傳異質(zhì)性的神經(jīng)發(fā)育障礙性疾病,常共患孤獨(dú)癥譜系障礙、注意缺陷多動(dòng)障礙等多種精神行為障礙,是全球兒童致殘的主要原因之一,其病因復(fù)雜,涉及遺傳和環(huán)境等多種因素[13]。近年來(lái),隨著基因檢測(cè)技術(shù)在臨床實(shí)驗(yàn)室的廣泛應(yīng)用,一些與嬰幼兒發(fā)育遲緩相關(guān)的基因備受關(guān)注。矮小癥是指與同地區(qū)、同年齡、同性別正常兒童比較,身高位于正常兒童生長(zhǎng)曲線的第3百分位數(shù)以下[14]。有諸多原因會(huì)影響兒童生長(zhǎng),導(dǎo)致兒童矮小,如遺傳、營(yíng)養(yǎng)、環(huán)境、精神心理因素、下丘腦-垂體-胰島素樣生長(zhǎng)因子軸異常、染色體畸變、全身性慢性疾病、遺傳代謝病以及內(nèi)分泌因素等。矮小合并畸形或發(fā)育異常者需要警惕遺傳代謝性疾病[15]。
本研究2例GRIN1基因突變引發(fā)發(fā)育遲緩的患兒中有1例同時(shí)合并矮小癥。GRIN1基因突變引發(fā)發(fā)育遲緩極為罕見(jiàn),目前僅報(bào)道30余例,尚未見(jiàn)合并矮小癥的報(bào)道。結(jié)合文獻(xiàn)報(bào)道,包括本研究2例患兒在內(nèi)的39例患兒的共同臨床特征為發(fā)育遲緩、認(rèn)知障礙、不同程度的智力受損等[4-12]。多數(shù)[72.2%(26/36)]患兒無(wú)自行行走能力,本研究病例1有此表現(xiàn)。部分患兒還表現(xiàn)為刻板運(yùn)動(dòng)[2.6%(1/39)]和眼動(dòng)障礙[12.8%(5/36)],本研究2例患兒均無(wú)此表現(xiàn)。其他臨床表現(xiàn)還有:孤獨(dú)癥譜系障礙[20.5%(8/39)]、性格激進(jìn)[2.6%(1/39)]、自殘行為[2.6%(1/39)]、痛覺(jué)異常[2.6%(1/39)]、睡眠障礙[20.5%(8/39)]、喂養(yǎng)困難[23.1%(9/39)]、小頭畸形[2.6%(1/39)]等,其中畸形是非特異性的臨床特征[6-7]。本研究2例患兒均無(wú)以上表現(xiàn)。
GRIN1基因突變患者中大部分有癲癇史[43.6%(17/39)]。癲癇發(fā)作表現(xiàn)為多種類(lèi)型,如全身性癲癇、局灶性癲癇、嬰兒痙攣、強(qiáng)直癲癇、肌陣攣性癲癇和發(fā)熱性癲癇等,癲癇發(fā)作時(shí)的年齡、腦電圖類(lèi)型和抗癲癇治療效果有明顯差異[6-7,9-11]。本研究2例患兒在隨訪期內(nèi)均無(wú)癲癇發(fā)作,這與文獻(xiàn)報(bào)道(突變位點(diǎn)相近的患兒無(wú)癲癇發(fā)作)[7]一致。考慮到GRIN1基因突變患兒癲癇初次發(fā)作的年齡有較大差異,本研究中有1例患兒年齡尚小,因此仍需長(zhǎng)期隨訪。
N-甲基-D-天冬氨酸受體(N-methyl-D-aspartate receptor,NMDAR)在正常的大腦發(fā)育和功能中起著重要作用。NMDAR亞單位的突變與多種癲癇綜合征及一些神經(jīng)發(fā)育障礙相關(guān)。由GRIN1基因編碼的NMDAR NR1亞基包含4個(gè)結(jié)構(gòu)域:(1)細(xì)胞外氨基末端結(jié)構(gòu)域,主要調(diào)節(jié)離子通道開(kāi)放和反應(yīng)時(shí)間過(guò)程;(2)ATD;(3)具有4個(gè)疏水段(M1~M4)的TMD;(4)CTD,參與定位和細(xì)胞內(nèi)信號(hào)轉(zhuǎn)導(dǎo)[1]。NR1亞基的激活涉及幾個(gè)調(diào)節(jié)因素機(jī)制。NR1亞基上存在NMDAR激動(dòng)劑之一的甘氨酸(glycine,Gly)結(jié)合位點(diǎn)。NR2亞基上的Glu結(jié)合位點(diǎn)通過(guò)結(jié)合Glu觸發(fā)調(diào)節(jié)向內(nèi)電流陽(yáng)離子選擇孔的打開(kāi),從而導(dǎo)致神經(jīng)元去極化和細(xì)胞內(nèi)鈣離子流入增加[16]。NMDAR的激活需要激動(dòng)劑Gly的輔助。NR1亞基與NR2亞基結(jié)合后才能形成具有高度活性功能的NMDA通道,調(diào)節(jié)鈣離子的內(nèi)流。NMDAR也有幾種負(fù)調(diào)節(jié)方式,包括電壓依賴型細(xì)胞外鎂離子、細(xì)胞外離子及內(nèi)源性細(xì)胞外鋅離子[1]。
本研究和文獻(xiàn)報(bào)道的39例患兒中,GRIN1基因突變復(fù)合雜合變異32例、純合變異7例。患兒預(yù)后:有3例死亡,死亡患兒出生后即出現(xiàn)難以控制的癲癇,死亡時(shí)年齡較小,其他36例表現(xiàn)為不同程度的智力發(fā)育遲滯、運(yùn)動(dòng)障礙等。
大多數(shù)GRIN1基因突變靠近或集中于構(gòu)成固有離子通道的4個(gè)TMD受體的區(qū)域(圖2)。TMD區(qū)域的突變與離子孔破裂有關(guān),表現(xiàn)為功能失調(diào)或缺乏功能[4]。本研究病例1發(fā)現(xiàn)的新突變p.Phe558Val在連接配體結(jié)合區(qū)(ligandbinding domain,LBD)和TMD的前M1區(qū),可能在傳遞配體信息中起作用,誘導(dǎo)LBD到TMD的結(jié)構(gòu)變化。因此,p.Phe558Val突變可能影響NMDAR的功能,進(jìn)而出現(xiàn)神經(jīng)系統(tǒng)損傷的臨床表現(xiàn)。病例2的p.Gly618Ser變異位于跨膜區(qū),對(duì)于p.Gly618和p.Gly620位點(diǎn)的突變,有模型預(yù)測(cè)可能會(huì)影響Glu NR1亞基順利進(jìn)入對(duì)離子的選擇性通過(guò)起決定作用的通道。因此,孔殘基p.Gly618和p.Gly620轉(zhuǎn)化為帶電精氨酸側(cè)鏈可能對(duì)NMDA通道屬性有很大的影響[8]。NMDAR的NR1亞基出現(xiàn)突變,會(huì)影響神經(jīng)元去極化和細(xì)胞內(nèi)鈣離子的流入超載,導(dǎo)致相關(guān)神經(jīng)元功能損傷。
有研究結(jié)果顯示,在僅表達(dá)Glu NR1正常水平5%~10%的GRIN1突變亞型小鼠模型中,小鼠的刻板行為增加,具有R844C純和突變的GRIN1突變小鼠模型具有多動(dòng)、社會(huì)互動(dòng)減少、驚嚇?lè)磻?yīng)及異常的焦慮樣行為[17-18]。盡管這些小鼠模型的行為異常與本研究的雜合子患兒不完全相似,但表明GRIN1基因突變可能在2個(gè)物種的社會(huì)和認(rèn)知行為中起著相似的作用。
GRIN1突變具有相似的臨床特征。但PADEROVA等[12]報(bào)道的1例GRIN1基因突變的9歲男孩,突變位點(diǎn)為c2470A>T,患兒有嚴(yán)重的智力發(fā)育延遲和運(yùn)動(dòng)遲緩,肌張力減退、輕度小頭畸形、小指的單側(cè)畸形、短手指、手指墊突出、關(guān)節(jié)松弛,還具有特殊面容:長(zhǎng)眼瞼、眼瞼下垂、藍(lán)鞏膜、斜視、高而窄顎弓、尖下巴、大耳、低位耳、耳廓畸形。這些臨床特征與歌舞伎綜合征類(lèi)似,但缺乏歌舞伎綜合征的分子基因診斷。其他NMDAR亞單位突變也有交叉重疊的臨床表現(xiàn),如GRIN2A基因突變與語(yǔ)言異常及一系列的癲癇性腦病有關(guān)。有研究結(jié)果顯示,已知基因中致病性變異的表型表達(dá)可能與不同的遺傳模式有關(guān)。目前尚未見(jiàn)編碼NMDAR其他亞單位(如GRIN2A)基因的純合或雙等位變異的報(bào)道[19]。
綜上所述,本研究發(fā)現(xiàn)了2例新的GRIN1基因突變引發(fā)的發(fā)育遲緩患兒,擴(kuò)展了該基因變異的基因型和表型庫(kù)。對(duì)于發(fā)育遲緩的患兒,排除繼發(fā)因素后,應(yīng)重視GRIN1基因突變的檢測(cè)。完善該類(lèi)患兒的基因檢測(cè),有助于明確病因,精準(zhǔn)診斷,同時(shí)也有助于患兒及其父母的遺傳咨詢,做好產(chǎn)前診斷工作。
猜你喜歡
英語(yǔ)世界(2023年6期)2023-06-30 06:29:10
中國(guó)民間療法(2021年5期)2021-06-09 09:21:04
中國(guó)生殖健康(2020年2期)2021-01-18 02:51:26
小學(xué)生導(dǎo)刊(2018年13期)2018-06-29 03:49:00
飲食科學(xué)(2017年5期)2017-05-20 17:11:53
雜文選刊(2016年7期)2016-08-02 08:39:56
小天使·一年級(jí)語(yǔ)數(shù)英綜合(2016年6期)2016-05-14 12:21:05
西南軍醫(yī)(2015年4期)2015-01-23 01:19:30
中國(guó)中醫(yī)藥現(xiàn)代遠(yuǎn)程教育(2014年20期)2014-03-01 04:31:21
河南醫(yī)學(xué)研究(2014年5期)2014-02-27 14:52:41
