連續微反應加氫技術在脫保護反應中的應用
2021-03-06 02:58:02婁鋒炎尹佳濱段笑南王祁寧艾寧張吉松
化工學報 2021年2期
關鍵詞:催化劑
婁鋒炎,尹佳濱,段笑南,王祁寧,艾寧,張吉松
(1 浙江省生物燃料利用技術研究重點實驗室,浙江杭州310014; 2 化學工程聯合國家重點實驗室,清華大學化學工程系,北京100084; 3 浙江工業大學化學工程學院,浙江杭州310014; 4 嘉興學院生物與化學工程學院,浙江嘉興314001)
引 言
在醫藥與精細化工領域中,保護與脫保護是一種較為常見的有機合成策略。多官能團底物進行多步有機合成時,通常需在反應活性位引入相應保護基以避免副產物的生成。常見的保護基主要有芐基和芐氧羰基等。其中N?和O?芐基是有機合成中最常用的保護基[1],一般可通過芐基鹵取代或苯甲醛縮合反應將其引入底物分子中[2],用于保護醇、酚、羧酸和酰胺等物質[3],使氨基、羥基等敏感基團在多步合成過程中保持穩定,而后根據產品要求進行芐基脫保護。芐氧羰基是一種常見的胺保護基,又稱Cbz[4],主要用于合成多肽。其與芐基保護基均可通過催化加氫、催化氫轉移氫解[5?8]、均相還原反應[9]或酶促法[10]等方式實現快速而高選擇性地脫保護。當底物分子無酸敏感基團時,還可通過對甲苯磺酸[11]、HBr/HOAc、液態HBr、液態HF[12]和三氟乙酸/茴香硫醚[13]等均相酸實現芐基或芐氧羰基的脫保護。其中,催化氫轉移氫解和均相還原法具有反應條件溫和、選擇性高和操作簡單等優勢,但存在成本較高、廢液產生較多等問題;酶促法面臨的菌種篩選和培育問題制約其發展;而催化氫解憑借其原子經濟性與高效綠色化等優點在醫藥中間體等有機合成中得到廣泛應用。莫西沙星[14?15]、美羅培南[16]和多利培南[17]等抗菌藥的合成過程中均有涉及催化加氫脫保護。
傳統的催化加氫脫保護反應[16,18?21]一般選擇配有攪拌槳的高壓間歇加氫釜為反應器,氫氣以氣泡形式引入反應體系。攪拌可增強氣液兩相的湍動程度,增大催化劑表面物質的更新速率;同時破碎氣泡使氣液兩相界面積增大,并延長氫氣在液相中的停留時間以增加氫氣的溶解量,強化傳質和傳熱效率,但依然存在氣液傳質效率低、反應時間長等問題,有些較難脫除的芐基甚至需要72 h 才能完成[22]。連續微反應加氫技術是連續流動化學與微反應器技術的結合體,它的出現為實現高效、綠色且可持續的有機化學合成提供了可能[23]。該技術利用微通道的優勢增加氣液固三相界面接觸面積,極大強化了多相傳質和傳熱,并顯著縮短反應時間到分鐘級甚至秒級。目前連續流加氫反應器主要有壁載式、填充式和漿料式三種路線[24]。文獻中報道的連續流脫保護反應基本選擇填充式連續流微反應器(圖1),其軸向返混通常較小,使氣液兩相的停留時間分布較窄,可減少連串副反應[25]。其次,由于微反應器體積較小,其氫氣滯留量相對較少[26],且催化劑因固載化而無須分離,操作安全性較高。近年來,連續流非均相加氫技術因其安全、高效等特點已得到越來越多的關注。不少國內外學者參與該技術的研究與討論,并在有關綜述[24,26?27]中介紹連續微反應加氫技術在N?、O?芐基和N?Cbz 脫保護方面的應用及其顯著優勢,包括產物的高選擇性和較短的反應時間。
本文首先簡要介紹了加氫脫保護中催化劑與溶劑的選擇,而后介紹了連續流非均相加氫技術在脫保護領域中的應用,尤其是在藥物中間體的合成領域,最后對該技術在脫保護中的應用進行了展望。

圖1 用于加氫脫保護的連續微填充床反應器示意圖Fig.1 Schematic diagram of continuous micro packed bed reactor for hydrodeprotection
1 催化劑與溶劑的選擇
1.1 催化劑對加氫脫保護反應的影響
催化劑對非均相催化加氫脫保護工藝至關重要,尤其是在連續化生產過程中,固體催化劑的穩定性對整個工藝過程有很大影響[28]。其中較為常見的加氫脫保護催化劑如表1所示。
相比于金屬鎳、貴金屬鉑、銠和釕,金屬鈀因吸氫性能好[42]、催化活性高、產物選擇性好等優勢已成為主流的脫芐基催化劑,也是目前芐氧羰基脫保護的首選催化劑。考慮其成本較高,通常選擇比表面積較大的多孔活性炭或氧化鋁為催化劑載體,使其催化活性位點得到充分利用。其中活性炭對金屬鈀的作用力較弱,可減小其對催化劑活性的影響,使Pd/C催化劑具有較好的催化活性[43]。

表1 常見的加氫脫保護催化劑、溶劑和添加劑Table 1 Common hydrogenation deprotection catalysts,solvents and additives
催化劑的加氫活性還與載體孔徑分布有關。當底物分子較大時,其無法擴散至載體的微孔和部分中孔[32],且可能堵塞載體孔道,減少催化活性中心的有效利用率。此時,要求載體具有較大的孔徑與孔容,使大分子底物能夠更好地接觸催化活性位。陳莉[16]采用介孔活性炭和普通活性炭負載的鈀催化劑進行保護美羅培南的脫芐基/芐氧羰基,研究結果表明前者的加氫活性與穩定性皆明顯優于后者,這主要是由于普通活性炭載體上的部分微孔或中孔被大分子副產物阻塞。李岳鋒等[44]采用Na2CO3調節催化劑前體H2PdCl4水溶液的pH,使之形成體積較大的鈀螯合離子,并借助位阻效應和活性炭表面的豐富基團,使鈀螯合離子在浸漬的過程中均勻吸附于活性炭外表面,形成“蛋殼形”Pd/C 催化劑以氫解保護美羅培南。該制備方法既可滿足反應物與催化劑活性位的良好吸附,又可消除內部傳質阻力的影響。
氫氧化鈀含量為20%的Pearlman 催化劑是一種常用于脫芐基的活潑催化劑[1],當Pd/C 催化劑對脫N?芐基失效時,該催化劑仍可顯現優越的性能[45]。Bernotas 等[46]還發現該催化劑可選擇性使胺加氫脫N?芐基而使芐基醚保持穩定。其認為含胺的底物可有效除去促進芐基醚氫解的微量酸,從而阻礙或抑制芐基醚的脫保護。但由于Pearlman 催化劑的活性組分負載量較高,導致其生產成本較高,一定程度抑制了其應用。Li 等[47]利用1∶1 的Pd/C 和Pd(OH)2/C 混合催化劑對多種脂肪醚/胺與芳香醚/胺進行加氫脫保護,發現等質量的混合催化劑的催化效率較單一的Pd/C 或Pd(OH)2/C 要高,反應時間也可縮減一半左右,并能有效解決Pd(OH)2/C催化劑的成本問題。除Pd/C、Pd(OH)2/C等高活性氫解催化劑外,貴金屬/樹脂催化劑的研究也得到廣泛關注。Dong 等[36]在室溫、常壓條件下,將10% Pd/C 替換為Pd/Dowex,并利用鈀與酸性樹脂的協同作用對四乙酰基二芐基六氮雜異伍茲烷進行N?芐基脫保護,產物的收率由92%提升至98%,并減少了貴金屬鈀的負載量。Koskin 等[48]推測HBIW 氫解期間的催化劑失活由低聚產物堵塞活性炭孔道和金屬顆粒團聚引起。Fotouhi?Far 等[49]進一步研究發現催化劑活性位周圍的pH 下降會導致部分金屬鈀晶粒浸入液相,導致催化劑負載量減少,且鈀晶粒的團聚將顯著減小催化劑比表面積,削弱催化劑對H2的化學吸附作用,從而影響其加氫活性。Maksimowski等[50]為解決HBIW 脫芐基時的Pd/C 失活問題,開發了一種催化劑再生工藝,即在5 L/h 的氮氣/水蒸氣流中,將失活的催化劑加熱至350℃,并持續2 h。使用再生的催化劑進行HBIW 加氫脫芐基,產物收率約為42%。
1.2 溶劑對加氫脫保護反應的影響
溶劑對非均相催化加氫體系的影響也較大,通常涉及氫氣的溶解度,溶劑分子在催化劑活性位上的競爭性吸附,催化劑顆粒的團聚以及反應物或產物分子與溶劑之間的相互作用[51]。不同催化劑?溶劑組合的加氫活性略有差異,其中較為常見的加氫脫保護溶劑如表1所示。
連續流催化加氫脫保護通常選擇低級醇、四氫呋喃、乙酸乙酯、乙酸和二甲基甲酰胺等極性溶劑為反應溶劑,其中二甲基酰胺在含OH?的水溶液中會逐漸分解為甲酸根與二甲胺[52],而游離胺的存在會影響催化劑的部分活性。鑒于甲醇和乙醇的低毒性、低成本和高溶解性,其通常被視為最佳溶劑。Pandarus 等[40]選取SiliaCat Pd(0)為催化劑,并以甲醇、乙醇或四氫呋喃為反應溶劑分別進行1?(芐氧基)?4?甲氧基苯的加氫脫保護。研究結果表明,底物在甲醇中的氫解速率最快,且底物濃度過高會減緩甚至抑制氫解反應。
脫保護溶劑還可分為單相和雙相溶劑,后者有助于實現氫解產物與保護基的相分離,可有效解決目標產物的分離問題[17],并避免因有機組分快速固化而造成管線堵塞[29]。Perosa 等[34]分別利用純乙醇和KOH?異辛烷?Aliquat?336 混合溶劑進行芐甲基醚的氫解,發現Pd/C 催化劑在乙醇中的氫解效率明顯較高,而Raney?Ni 恰恰相反。Perosa 等推測催化劑表面涂覆的Aliquat?336 可促進Raney?Ni 向反應的有機相轉移,并為底物提供具有特定官能團催化位點的反應微環境。
David 等[19]為研究體系pH 與脫芐基和脫氯的選擇性關系,以4?氯?N,N?二芐基苯胺為底物,并采用三乙胺/乙酸緩沖液調節反應體系的pH。研究結果表明,當體系的pH 高于被保護胺的pKa時,可以實現高選擇性脫氯;反之,有機胺將以質子化形式存在,增強N?Bn 鍵(Bn 代表芐基)的親電性,既可抑制脫氯的部分活性,又能防止催化劑失活。
本課題組[33]在連續氫解芐基類保護基時發現四氫呋喃和2?甲基四氫呋喃對Pd(OH)2/Al2O3催化劑具有一定的毒害作用,可使氫解轉化率緩慢下降,但具體的催化劑失活機理有待進一步研究。
此外,氫氣在傳統有機溶劑中的溶解度一般較低,但與超臨界流體(如超臨界二氧化碳)完全混溶。超臨界流體憑借其低黏性、良好的傳質和傳熱性能以及易與產物和溶劑相分離,將在連續催化加氫領域占據重要地位[53]。
2 連續微反應加氫脫保護
2.1 脫芐基反應
2.1.1 O?芐基脫保護 O?芐基是藥物合成過程中的常用保護基,可用于保護酚、醇和羧酸類有機化合物。在有關藥物化學的文獻資料中,O?芐基的脫除占所有脫保護反應的1.4%[54]。多羥基化合物的合成過程中,各保護基的氫解順序會嚴重影響加氫產物的選擇性。因此,需要不同反應活性的芐基保護基實現正交保護基策略[55]。Gaunt 等[55]為此展開相關研究,發現不同芐基醚的氫解順序由取代芐基芳環上的電子效應和底物對金屬鈀表面的親和力控制。芐基的芳環連有供電子基時,其通常較易被脫除;反之,則較為穩定。然而芳環取代基的存在可能誘發不利的空間效應并影響鍵合所需的平面幾何結構,從而降低氫解活性[56]。
Sajiki 等[57?58]發現游離胺可能會抑制脂肪族/芳香族芐基醚的氫解,并考察了鄰位取代的吡啶衍生物對脂肪族芐基醚加氫脫保護的抑制作用,發現吡啶環上的氮孤對電子周圍的空間位阻可以減弱該含氮堿與金屬鈀的相互作用(其對鈀表面的親和力和對鈀表面活性位點的抑制作用),從而可能避免Pd/C 催化劑的失活;其次,含氮堿的兩氮原子之間的C—C 鏈長度在抑制芳香族芐基醚的氫解反應中起至關重要的作用。Yin 等[59]在優化雷貝巴坦(relebactam)關鍵羥胺中間體的合成路線時發現三乙胺和DABCO 可顯著提高芐基醚的氫解速率,而2,6?二叔丁基吡啶對該反應具有抑制作用,這與上述Sajiki等的說法不一致。
此外,多數用于芐基醚氫解的催化劑也可能導致芐基芳環的飽和。Crawford 等[60]在氫解芐基醚和萘甲基醚期間發現樣品中含有部分飽和醚,這無疑增大了產物的提純難度。Ochocinska 等[61]以10%Pd/C 為催化劑,DMF?HCl 水溶液為溶劑,成功解決了萘甲氧基糖苷的氫解選擇性問題,而無芳環飽和或酸不穩定基團分解等副反應。
連續流微反應器憑借其高傳質和傳熱效率以及顯著較短的反應時間被廣泛用于脫保護反應條件的快速篩選和優化以及氫解產物的高通量合成。
Knudsen 等[62]利用H?Cube 反應器仔細研究了反應溫度、底物流量和濃度對Boc?O?Bn?L?酪氨酸氫解的影響,發現增加底物的流量與濃度可加速催化劑失活,且溫度的上升對維持催化劑活性有顯著影響。在60℃的反應條件下,經過連續7 次加氫脫保護,底物的轉化率仍接近100%(圖2)。

圖2 N?Boc?O?芐基?酪氨酸的連續氫解[62]Fig.2 Continuous hydrogenolysis of O?benzyl protected N?Boc?tyrosine[62]
Ekholm 等[29]在連續流加氫微反應器中以Pd/C為催化劑,MeOH?EtOAc 為混合溶劑,并通過高壓柱塞泵將1 mg/ml的(3R,4R,6S)?3,4,5?三(芐氧基)?2?[(芐氧基)甲基]?6?甲氧基氧烷溶液以1 ml/min的流量送入系統進行芐基醚脫保護處理,產物甲基?α?D?吡喃甘露糖苷的收率高達95%(圖3)。

圖3 甲基?α?D?吡喃甘露糖苷的連續合成[29]Fig.3 Continuous synthesis of methyl?α?D?mannopyranoside[29](1bar=0.1 MPa)
Desai 等[63]在H?Cube 反應器中填充10% Pd/C,對含有酰胺鍵、碳碳雙鍵的DHPM 芐基酯1a~1d 進行芐酯脫保護以獲得相應的DHPM 酸(表2),為后續3,4?二氫嘧啶?2?酮衍生物(可用于制備抗高血壓藥和鈣拮抗劑)的高通量合成奠定了基礎(圖4)。

圖4 DHPM芐基酯的連續氫解[63]Fig.4 Continuous hydrogenolysis of DHPM benzyl esters[63]

表2 連續流條件下四種DHPM 芐基酯的氫解Table 2 Hydrogenolysis of four DHPM benzyl esters under continuous flow conditions
Zhang等[64]使用H?Cube反應器對四氫萘酰胺衍生物(表3 a~d)和二氫茚酰胺衍生物(表3 e~g)進行芐基醚脫保護(圖5~圖6)。當氫氣壓力為0.1 MPa、反應溫度為45℃時,前者的收率可達86%以上,后者在2 MPa 的氫氣壓力下也可獲得86%以上的高收率。

圖5 四氫萘酰胺類衍生物的連續氫解[64]Fig.5 Continuous hydrogenolysis of tetrahydronaphthalene amide derivatives[64]

圖6 二氫茚酰胺類衍生物的連續氫解[64]Fig.6 Continuous hydrogenolysis of dihydroindene amide derivatives[64]
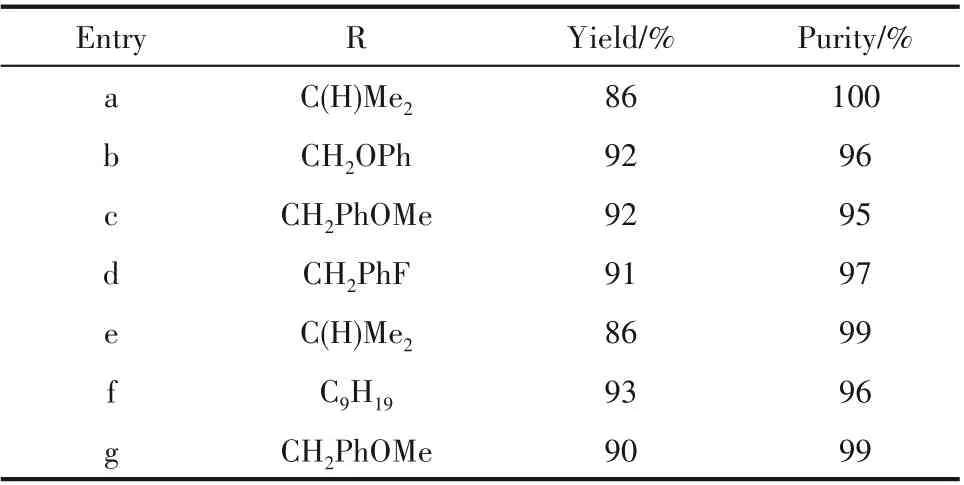
表3 雙環羥基酰胺的合成Table 3 Synthesis of bicyclic hydroxy amides
2.1.2 N?芐基脫保護 芐基容易引入,但較難脫除,特別是N?芐基的脫保護[65?66]。相比于O?芐基加氫脫保護,其氫解過程通常需要更高的催化劑負載量、反應溫度和壓力[67]。受N?芐基保護的叔胺和季胺一般在室溫與常壓下較易被氫解,而伯胺或仲胺需要更高的壓力(>4 bar)和溫度(>40℃)[68]。同時N?芐基氫解產生的游離胺會與催化劑的活性中心產生強相互作用,從而削弱催化劑的部分活性甚至使其完全失效[42]。此外,Tanielyan 等[69]意外發現低壓氫氣有利于提高含吡啶酮和芳香氟化物結構的復雜芐胺的N?芐基脫保護速率,即提高產物的選擇性。一種可能性是使用低壓氫氣可減小催化劑表面的氫氣覆蓋率,從而提高芐基在催化劑表面的吸附程度,加快脫芐基反應。
Jones 等[70]在H?Cube 連續流反應器中填充10%Pd/C催化劑,并以EtOAc/EtOH(1∶1)為混合溶劑,對N?芐基?2?苯乙胺進行氫解,實現了100%的轉化率和89%的收率(圖7)。該連續流反應體系有效解決了N?芐基脫保護問題,并為Boc 和Fmoc 保護基的酸解提供了有效替代方案。
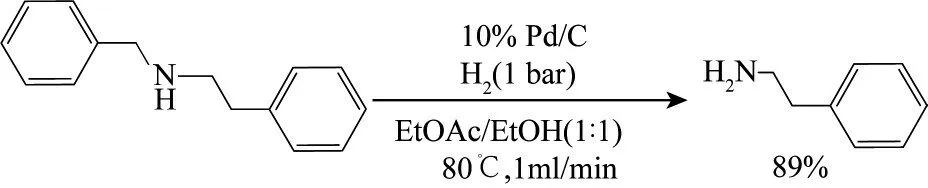
圖7 N?芐基?2?苯乙胺的連續氫解[70]Fig.7 Continuous hydrogenolysis of N?benzyl?2?phenylethylamine[70]
Darvas 等[71]使用實驗室規模的連續流加氫裝置對N?芐基?2?苯乙胺的脫保護也進行相關研究。其以EtOAc/EtOH 為溶劑,鈀黑為催化劑,當底物濃度為0.05 mol/L 時,苯乙胺收率將近99.9%。相比于Jones等[70]的實驗結果,其收率有顯著提高(圖8)。

圖8 N?芐基?2?苯乙胺的連續氫解[71]Fig.8 Continuous hydrogenolysis of N?benzyl?2?phenylethylamine[71]
Baxendale 等[72]使用HEL 連續流加氫系統,并耦合微波和連續流技術,進行1?芐基?1,4,5,6?四氫環戊并[b]吡咯?2?羧酸乙酯的加氫脫保護,同時飽和其芳環生成相應吡咯烷作為雷米普利前體,其產物收率可達97%(圖9)。
本課題組[33]采用微填充床反應器成功實現了N?二苯甲基的連續高效氫解脫除。該系統經過190 h 的連續運行,產物收率仍可維持在99.5%以上。此外,對比了傳統間歇式反應器(250 ml)和微填充床反應器在相同反應溫度和壓力下的反應性能,發現后者單位體積單位時間的原料轉化量是前者的100 倍,這極大地顯示了連續流微反應器的氫解優勢。

圖9 1?芐基?1,4,5,6?四氫環戊并[b]吡咯?2?羧酸乙酯的連續氫解[72]Fig.9 Continuous hydrogenolysis of ethyl 1?benzyl?1,4,5,6?tetrahydrocyclopenta[b]pyrrole?2?carboxylate[72]
2.2 脫芐氧羰基反應
芐氧羰基常用于保護胺類物質,一般出現于多肽的合成過程中。相比于脫芐基,其氫解產物還包括CO2。Müslehiddino?lu 等[73]在間歇加氫反應釜中以Pd/C 為催化劑,對N?芐氧羰基的氫解過程進行了研究。結果表明,CO2對催化劑的影響較大,其會改變脫保護反應的速率與級數。此外,他們分別利用碳酸鉀和三乙胺進行CO2的吸收與溶液pH 的調節,發現兩種方式均可降低催化劑的中毒程度,同時借助Langmuir?Hinshelwood 機理描述芐氧羰基的氫解過程,提出產物中間體的形成步驟為決速步驟。而戴云生等[42]表示N?芐氧羰基型保護基的氫解體系不局限于堿性環境,也可為中性或酸性,后者可能是利用有機胺的質子化,從而降低催化劑的中毒程度。黃建珍[28]利用填充床振蕩流反應器合成氨曲南主環,并借助裝置的平推流與無梯度特性將生成的CO2迅速帶走,使氫氣分壓保持恒定以探究反應的宏觀動力學。其同樣認為產物中間體的形成步驟為決速步驟。此外,Gaunt等[74]發現富電子的2?萘甲基氨基甲酸酯的氫解活性遠不及芐氧羰基,這與此前該團隊研究的2?萘甲基?芐基體系形成鮮明對比。其推測這可能與N?芐氧羰基的自身反應性較強有關。
Oyamada 等[35]將高活性、低浸出率、無溶脹的Pd/(PSi?Al2O3)催化劑運用于連續流動反應器中,并可能考慮到Cbz?L?絲氨酸易溶于醇溶劑,產物絲氨酸易溶于水,而選擇乙醇/水(1∶4)為混合溶劑使整個反應在溶液中進行,最終成功實現Cbz?L?絲氨酸的加氫脫保護[圖10(a)]。此外,該團隊發現Pd/(PSi?Al2O3)催化劑在此氫解系統中可至少維持8 h的高活性,且在反應期間無金屬鈀浸出。

圖10 N?芐氧羰基的連續氫解[35,70?71,75,78?79]Fig.10 Continuous hydrogenolysis of N?benzyloxycarbonyl[35,70?71,75,78?79]
Jones 等[70]在H?Cube 反應器中填充10%Pd/C 催化劑,并在常壓、50℃的反應條件下,對含雙鍵結構的(2?(1H?吲哚?3?基)乙基)氨基甲酸芐酯進行N?芐氧羰基脫保護,最終獲得較高收率的色胺[圖10(b)]。Darvas 等[71]利用實驗室規模的連續流加氫裝置對0.05 mol/L 的上述底物溶液也展開了相應脫保護研究,相比于Jones 等的實驗結果,其產物收率有顯著提高,可達95%[圖10(c)]。
苯乙胺是制備藥物或染料的重要中間體,主要用于合成抗抑郁藥、支氣管擴張藥等。Kobayashi等[75?77]利用聚合物包埋法將鈀絡合物固載于玻璃微通道內壁,同時要求氣?液兩相以環流形式與催化劑相接觸,在1 min 內成功實現苯乙基氨基甲酸芐酯的N?Cbz 脫保護,且產物收率接近100%[圖10(d)]。
Ladlow 等[78]利用H?Cube 反應器依次對預先合成的Cbz?L?Val?L?Phe?OMe 等肽鏈進行連續流氫解[圖10(e)]。為解決較長肽鏈的溶解性問題,該團隊選擇二甲基甲酰胺為氫解溶劑。實驗結果表明,受芐氧羰基保護的較長肽鏈在60℃和0.1 MPa 下均可獲得不同程度的脫保護,這為將來的藥物和多肽合成提供了新的可能。
Clapham 等[79]設計、構建了一套自動化連續流加氫裝置以快速篩選與優化2?[(4?苯氧基苯基)氨基甲酰基]吡咯烷?1?羧酸芐酯的脫保護反應條件,并借助液質聯用分別對30、40、60 和80℃反應溫度下的樣品進行檢測,發現在60℃、0.1 MPa 的反應條件下該氫解反應可得到100%的轉化率[圖10(f)]。
3 結論與展望
連續微反應加氫技術是一種原子經濟且高效綠色化的合成手段。相較于高壓間歇加氫釜等傳統反應器,連續流微反應器可利用其微通道優勢解決氣?液?固三相界面接觸面積小、相間傳遞速率低等問題,其高效的氣液傳質效率與平推流特性還可實現高選擇性脫保護,并顯著縮短反應時間。此外,加氫脫保護通常需在一定壓力下進行,以增加氫氣在溶劑中的溶解度及其在催化劑表面的吸附程度,故存在一定操作危險性,而連續微反應器憑借其體積小、持液量小、氫氣滯留量低等優勢可實現安全可控的生產目的。催化劑、溶劑和添加劑的合理選擇也可進一步提高氫解產率。Pd/C、Pd(OH)2/C 或兩者的混合物通常被視為高活性脫保護催化劑,且因固載化而無需分離,操作安全性較高;低級醇、四氫呋喃、乙酸乙酯、乙酸和二甲基甲酰胺等極性溶劑為常見的脫保護溶劑,而甲醇和乙醇因低毒性、低成本和高溶解性,通常被視為最佳溶劑;有機酸或含氮堿的引入可選擇性地促進或抑制芐基,從而提高產物選擇性;雙相混合溶劑的應用還可助于氫解產物與保護基的相分離,既能有效解決目標產物的分離問題,又能避免有機組分快速固化而帶來的管線堵塞問題。目前連續微反應加氫技術在脫保護反應中的報道大多基于實驗室規模的合成與研究,本課題組已經成功實現了連續流加氫微反應器的放大,并運用于脫芐基產品的噸級生產。該成果表明,連續微反應加氫技術具有過程安全性高、反應時間短和催化劑成本低等特點,相比傳統的釜式加氫具有明顯優勢。但同時,該技術還面臨過程易堵塞、工藝開發時間長等問題。相信隨著該技術的不斷進步,其在未來將被廣泛運用于醫藥和精細化工等諸多領域。
猜你喜歡
大自然探索(2023年7期)2023-11-14 13:08:06
石油石化綠色低碳(2019年6期)2019-02-13 09:39:01
石油石化綠色低碳(2019年6期)2019-01-14 01:16:22
智富時代(2018年3期)2018-06-11 16:10:44
浙江大學學報(工學版)(2016年11期)2016-06-05 09:21:04
Coco薇(2016年2期)2016-03-22 02:45:06
超硬材料工程(2016年1期)2016-02-28 22:20:04
中國資源綜合利用(2016年4期)2016-01-22 08:27:23
合成化學(2015年4期)2016-01-17 09:01:27
應用化工(2014年3期)2014-08-16 13:23:50
