雙環磺草酮的合成研究
2021-03-06 12:56:18楊輝斌
現代農藥 2021年1期
關鍵詞:除草劑
張 帆,梁 爽,孫 冰,李 斌,楊輝斌
(沈陽中化農藥化工研發有限公司,新農藥創制與開發國家重點實驗室,沈陽 110021)
對羥苯基丙酮酸雙氧化酶(HPPD)是植物質體醌和生育酚合成過程中的關鍵酶。抑制HPPD活性可導致質體醌和生育酚的正常合成途徑被阻斷,進而造成類胡蘿卜素的生物合成減少、光合作用鏈電子傳遞受阻,最終導致植物中葉綠素被破壞,出現葉片白化癥狀,甚至死亡。因此,HPPD是理想的除草劑靶點。HPPD抑制劑類除草劑既可在苗前使用,也可在苗后使用,并具有高效、低毒、抗性低、環境友好以及使用安全等特點。自20世紀90年代以來,該類除草劑已成為農藥化學研究領域的熱點[1]。目前已開發的HPPD類除草劑主要有硝磺草酮、異唑草酮和苯唑草酮等[2-4]。2018年,在全球作物農藥市場,除草劑的銷售額為246.08億美元,同比增長5.9%,占全球作物用農藥市場的42.7%,其中HPPD抑制劑類除草劑增幅達11.1%,是除草劑中同比增速最快的產品類型[5]。
雙環磺草酮屬于雙環辛烷類HPPD除草劑,其結構中有新穎的苯硫醚和雙環結構,化學名稱為3-(2-氯-4-甲基磺酰基苯甲酰基)-2-苯硫基雙環[3.2.1]辛-2-烯-4-酮。2001年,雙環磺草酮在日本登記并投放市場,商品名為Show-Ace,通過莖葉噴霧,用于水稻田和移栽水稻田一年生雜草的去除,有效劑量為168~252 g/hm2。雙環磺草酮是內吸傳導型除草劑,主要通過植物根莖部的吸收,導致新葉白化,故對螢藺、異型莎草、扁稈藨草、鴨舌草、雨久花、陌上菜、澤瀉、野慈姑、幼齡稗草、假稻、千金子等都具有較好的防效,尤其對螢藺、鴨舌草防效突出。雙環磺草酮持效期可達30~60 d,具有緩釋效果,此外,其還具有處理適期寬和持效性長的特點[6-9]。
目前,關于雙環磺草酮合成方法研究的文獻報道較少,僅有關于葛發祥等[10]以降冰片烯為原料合成關鍵中間體雙環[3.2.1]辛烷-2,4-二酮,進一步合成除草劑雙環磺草酮原藥的報道。筆者以2-氯-4-甲磺酰基苯甲酸為起始原料,依次經過酰氯化,酯化,重排,氯化,硫醚化5步反應合成雙環磺草酮,并對酰氯化,酯化,重排3個關鍵步驟的工藝參數進行了系統研究,合成路線見圖2。

圖2 雙環磺草酮的合成
1 反應條件及合成研究
1.1 2-氯-4-甲磺酰基苯甲酰氯(M-1)的合成
文獻中關于2-氯-4甲砜基苯甲酰氯的合成方法主要有2種。分別采用二氯亞砜或草酰氯作為酰氯化試劑[11-12],在不同的溶劑及溫度條件下制備酰氯,得到的酰氯經濃縮后直接用于酯化反應。結果見表1。

表1 2-氯-4-甲磺酰基苯甲酰氯反應條件
首先,嘗試以二氯亞砜為溶劑的酰化條件(序號1),但原料2-氯-4-甲磺酰基苯甲酸在二氯亞砜中的溶解度較小,隨著反應溫度升高和反應的進行,原料逐漸溶解,反應時間約為2 h;其次嘗試以二氯甲烷為溶劑,采用二氯亞砜為酰化劑,DMF為催化劑的體系進行反應。該體系反應進程十分緩慢,40℃過夜后仍有大量原料剩余(序號2);然后嘗試以二氯乙烷代替二氯甲烷為溶劑,二氯亞砜-DMF體系進行反應的效果也不太理想(序號3);最后嘗試以活性更高的草酰氯為酰化試劑,DMF為催化劑,二氯甲烷為溶劑的酰氯化體系。該體系具有極高的反應能力,在0.5 h左右,原料即可完全轉化成酰氯,經減壓脫溶后得到酰氯粗品,直接用于酯化反應。
1.2 中間體M-2的合成
該酯化反應采用經典的酰氯-二酮-三乙胺體系,以二氯甲烷為溶劑,考察了底物投料比對反應的影響,結果見表2。
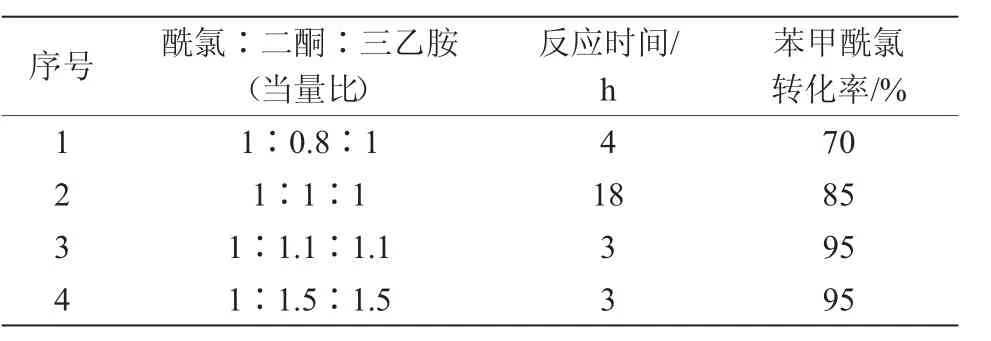
表2 酯化條件
當苯甲酰氯過量時(序號1),以三乙胺做堿化,室溫條件下反應4 h,雙環[3.2.1]辛烷-2,4-二酮轉化率可超過90%,但酰氯剩余較多,剩余的酰氯及苯甲酸需多次以飽和碳酸氫鈉溶液洗滌方可去除,操作繁瑣;將酰氯與二酮當量比變為1∶1時,即使過夜反應,2個原料仍有較多剩余(序號2);當二酮稍過量時,在相同的條件下,約3 h,酰氯即可實現較高的轉化率,過量的二酮經10%NaOH溶液洗滌1次即可去除(序號3);進一步地提高二酮當量對酰氯轉化率的提升無顯著影響(序號4)。最終筆者采用酰氯∶二酮∶三乙胺當量比1∶1.1∶1.1的條件,以極高的轉化率制備得到烯醇酯(M-2)。
1.3 中間體M-3的合成
文獻報道的烯醇酯重排條件為三乙胺-丙酮-氰化氫體系[6]。該體系由于需要通入氰化氫氣體,操作不便且危險性較大。三乙胺-丙酮氰醇體系是進行該重排反應常用方法。經實驗發現,重排反應的主要副反應是烯醇酯分解為2-氯-4-甲磺酰基苯甲酸與雙環[3.2.1]辛烷-2,4-二酮,結果見表3。
對該類反應的機制進行推測,如圖3所示。
由于氰離子極強的親核性,烯醇酯M-2很容易被氰離子進攻,形成高活性的苯酰腈中間體,若該中間體與體系中的水反應則生成苯甲酸副產物;若二酮以酮式進攻苯酰腈中間體則會形成穩定的重排產物M-3;若二酮的烯醇式進攻苯酰腈中間體則形成烯醇酯M-2,該M-2會繼續被氰離子進攻,進入新一輪催化循環,直至形成穩定的苯甲酸或重排產物。由反應機理可知,重排與水解為1對競爭反應,雖然二酮的親核性高于水,但在較高的溫度下,反應選擇性會降低。因此采用2種方案以避免副產物的形成:其一,降低體系中的水含量;其二,降低反應溫度以提高選擇性。最后采用無水硫酸鎂干燥二氯甲烷作為溶劑,在10~15℃條件下滴加丙酮氰醇,再緩慢升溫至室溫,攪拌過夜,獲得了較為滿意的反應效果。
2 實驗部分
2.1 儀器與試劑
主要試劑:所用試劑為市售化學純或分析純。
主要儀器:Mercury 300(Varian)核磁共振儀(TMS為內標),美國瓦里安有限公司;RY-1型熔點儀,天津分析儀器廠;Agilent 1100系列高效液相色譜,美國安捷倫公司。
2.2 關鍵中間體的合成步驟
2.2.1 2-氯-4-甲磺酰基苯甲酰氯(M-1)的合成
將2-氯-4-甲磺酰基苯甲酸1.30 g(5.55 mmol)、二氯甲烷30 mL加入到100 mL反應瓶中,加入1滴DMF,降溫至0℃,緩慢加入草酰氯1.41 g(11.1 mmol),滴畢,室溫攪拌0.5 h,減壓蒸除溶劑及過量的草酰氯,得白色固體(M-1)1.3 g,直接用于下一步反應。
2.2.2 4-氧代雙環[3.2.1]辛-2-烯-2-基2-氯-4-(甲磺酰基)苯甲酸酯(M-2)的合成
在250 mL反應瓶中加入雙環[3.2.1]辛烷-2,4-二酮0.84 g(6.08 mmol)、三乙胺0.62 g(6.12 mmol)以及二氯甲烷40 mL降溫至0℃,將上步制得的1.3 g M-1溶解在20 mL二氯甲烷中,緩慢滴加到反應瓶,加料完畢,室溫下攪拌3 h,減壓蒸除二氯甲烷,加入150 mL乙酸乙酯,以10%NaOH溶洗滌,有機相以飽和食鹽水洗滌至中性,干燥,濃縮,得白色固體(M-2)1.85 g,直接用于下一步反應。
2.2.3 3-(2-氯-4-甲基磺酰基苯甲酰基)-4-羥基-雙環[3.2.1]-2-辛烯-4-酮(M-3)的合成
往上步制得的1.85 g M-2中加入二氯甲烷40 mL、三乙胺0.85 g(8.40 mmol),降溫至10℃,攪拌下加入2~3滴丙酮氰醇,體系在3 h內緩慢升溫至室溫,室溫下攪拌15 h,反應液中加入1mol/L HCl溶液,用飽和食鹽水洗滌、干燥、減壓脫溶,殘余物經柱色譜提純(淋洗液為PE∶EA=1∶1)得到白色固體(M-3)1.12 g(3.16 mmol),三步收率為57%。1H NMR(300 MHz,CDCl3)δ:8.12-8.10(m,2H)、7.98-7.96(m,1H)、5.91(s,1H)、3.13(s,3H)、3.06-3.03(m,2H)、2.27(d,1H)、2.19-2.11(m,3H)、1.77-1.72(m,2H)。
2.2.4 3-(2-氯-4-甲基磺酰基苯甲酰基)-4-氯代雙環[3.2.1]-2-辛烯-4-酮(M-4)的合成
在100 mL的單口瓶中加入1.12 g M-3(3.16 mmol)和二氯甲烷30 mL,攪拌下加入草酰氯0.62 g(4.88 mmol)和2滴DMF,加料完畢,室溫攪拌4 h。減壓蒸出有機溶劑,殘余物加入50 mL乙酸乙酯,有機層依次用50 mL飽和碳酸氫鈉溶液、50 mL飽和食鹽水洗滌、無水硫酸鎂干燥,減壓脫溶,殘余物經柱色譜提純(淋洗液為PE∶EA=1∶1)得到白色固體(M-4)1.00 g,收率為89%。1H NMR(300 MHz,CDCl3)δ:7.96-7.78(m,3H)、3.06(s,3H)、2.35-1.72(m,8H)。
2.3 目標化合物雙環磺草酮的合成
在100 mL反應瓶中加入0.80 g M-4(2.15 mmol)和二氯甲烷50 mL,依次加入苯硫酚0.26 g(2.36 mmol)和三乙胺0.25 g(2.47 mmol),室溫攪拌4 h。反應液依次用50 mL飽和碳酸鈉溶液、50 mL飽和食鹽水洗滌、無水硫酸鎂干燥,減壓脫溶得黃色油液。向粗品中加入10 mL丙酮后,冷卻至0℃,保溫2 h,過濾,濾餅以丙酮洗滌,得到淡黃色固體(目標化合物)0.59 g,收率為61%。1H NMR(300 MHz,CDCl3)δ:7.92-7.49(m,8H)、3.09(s,3H)、2.18-2.06(m,2H)、1.87-1.78(m,4H)、1.59-1.56(m,2H)。13C NMR(75 MHz,CDCl3)δ:197.0、192.0、184.5、145.8、142.0、135.5、131.2、130.5、129.8、129.7、129.6、128.4、127.2、125.8、50.2、44.6、44.5、42.8、37.5、31.5、26.2。ESI-MS(m/z):447.0([M+H]+)、468.9([M+Na]+)。
3 結 論
筆者以2-氯-4-甲磺酰基苯甲酸為起始原料,經5步反應合成了雙環磺草酮,對關鍵中間體的合成參數進行了優化,縮短了反應時間,簡化操作,避免了副產物的生成。目標化合物的結構經1H NMR和MS確證。本研究對雙環磺草酮類衍生物的結構改造具有重要意義。
猜你喜歡
世界農藥(2019年3期)2019-09-10 07:04:10
今日農業(2019年15期)2019-01-03 12:11:33
現代園藝(2017年19期)2018-01-19 02:50:21
長江蔬菜(2016年10期)2016-12-01 03:05:27
獸醫導刊(2016年12期)2016-05-17 03:51:29
現代農業(2016年5期)2016-02-28 18:42:36
雜草學報(2015年2期)2016-01-04 14:58:05
種業導刊(2016年9期)2016-01-03 01:27:14
營銷界(2015年23期)2015-02-28 22:06:18
營銷界(2015年22期)2015-02-28 22:05:11
