毛竹水解渣木質素氫解制備芳香化學品
2021-03-05 08:22:34屈一新王際東
林產化學與工業 2021年1期
關鍵詞:質量
程 毅,屈一新,楊 昇,莊 抗,王際東*
(1.北京化工大學 化學工程學院,北京 100029;2.中國林業科學研究院木材工業研究所,北京 100091)
生物煉制是以生物質為原料,通過物理、化學或生物手段,獲得生物基燃料、材料或化學品的過程。木質纖維是一種儲量豐富且可再生的生物質資源,主要由纖維素、半纖維素和木質素組成[1]。典型的木質纖維生物煉制過程是先將原料的目標組分分離,再進一步轉化[2-3]。過程中產生的副產物往往被加工成低附加值產品或直接燃燒,難以得到高值化利用。優化纖維素、半纖維素和木質素的分離工藝,開發新型反應體系,實現全組分煉制,近年來受到越來越多學者的關注[4]。木質素可以通過化學或物理方法進行分離,進而實現各組分轉化。常見的木質素的化學分離方法包括過氧化氫法、有機溶劑法、堿法和亞硫酸鹽法等。于海龍[5]在對工業纖維渣脫木質素研究中發現:相同條件下,過氧化氫法分離糠醛渣中的木質素,效果優于含半纖維素的甘蔗渣;而有機溶劑法的結果正相反。這說明解除了半纖維素屏障的木質纖維,采用過氧化氫法分離木質素可以取得很好的效果。過氧化氫法分離木質素的過程中,脂肪族羥基被氧化成羰基或醛基,降低了木質素中氧醚鍵的解離能[6]。因此,過氧化氫法從戊糖水解渣中分離的木質素在化學品轉化方面擁有巨大潛力。過渡金屬催化劑,如Pd/C、Ru/C或Ni/C等,在供氫溶劑(甲醇、乙醇或四氫呋喃等)中轉化芳香化學品被證明是木質素轉化的一種有效方式[7-10]。為了防止降解產物發生再縮合,溶劑會采用供氫溶劑和水的混合體系,如甲醇/水或乙醇/水[11-13],提高目標產品的得率。本研究以毛竹提取半纖維素后的水解渣為原料,分別利用過氧化氫法和甲醇法提取木質素,以甲醇/水作為反應溶劑和氫源,Pd/C為催化劑,對兩種木質素進行氫解,獲得芳香化學品,使用GC-MS、FT-IR和GPC等手段對原料和氫解得到的產品結構進行分析,并據此對毛竹水解渣木質素氫解過程進行探索。
1 實 驗
1.1 材料、試劑與儀器
原料為三年生毛竹(Phyllostachysheterocyclacv.Pubescens),由國際竹藤中心提供。將竹節粉碎后篩分得到粒徑小于0.425 mm的樣品,平均含水率為9.3%,備用。絕干原料中纖維素、半纖維素和木質素質量分數分別為46%、24.9%和25%。
催化氫解所用的催化劑為Pd/C,Pd負載量為5%,美國Sigma-Aldrich公司。甲醇,分析純;FeCl3·6H2O,試劑純;四氫呋喃、甲醇和溴化鉀,均為光譜純。
Waters 8795e高效液相色譜(HPLC)儀,帶有示差檢測器(RID),美國Waters公司;色譜柱Aminex HPX-87P column (300 mm×7.8 mm),美國Bio-Rad公司;GC-2010 Plus氣相色譜-質譜聯用(GC-MS)儀,KB-WAX分離柱(0.25 mm×30 m×0.25 μm),日本島津公司;Tensor 27傅里葉變換紅外光譜(FT-IR)儀,德國布魯克公司;Waters 7890e凝膠滲透色譜(GPC),水相色譜柱PWxL Guard Column,TSKgel G3000PWxL和TSKgel G5000PWxL串聯,日本TOSOH公司;PLgel-Mixed-D四氫呋喃相色譜柱(300 mm×7.5 mm),美國Agilent公司。
1.2 毛竹水解渣的制備
參考文獻[14]的方法制備毛竹水解渣。取13.23 g原料(絕干物12 g)加入到200 mL高壓釜中,再加入4 g FeCl3·6H2O和120 mL水。攪拌下加熱至170 ℃保持35 min。反應結束后,將反應釜在冷水中急速冷卻至室溫,將混合物過濾分離。所得固體經水洗至水洗液為無色,在105 ℃烘箱中絕干4 h,即得到毛竹水解渣。
1.3 木質素的提取制備
1.3.1過氧化氫法 參考文獻[5]的方法,取0.75 g毛竹水解渣加入到25 mL聚四氟乙烯水熱釜中,加入14 mL pH值10.5的NaOH溶液(質量分數0.01%)和1 mL H2O2溶液(質量分數30%),將釜蓋迅速關閉擰緊;放入恒溫水浴鍋中,在磁力攪拌下于80 ℃反應4 h;然后,將反應釜在冷水中急速冷卻至室溫,過夜。反應結束后,過濾,將得到的處理液經真空旋轉蒸發濃縮至5 mL,處理液從淺黃色變成深棕色,將該深棕色液體逐滴加入到50 mL 95%乙醇中,所得懸濁液在室溫下靜置過夜后過濾,濾餅在空氣中風干,即可得到過氧化氫木質素。
1.3.2甲醇法 參考文獻[5]的方法,取1.5 g毛竹水解渣加入到25 mL聚四氟乙烯水熱釜中,再加入7.5 mL的甲醇和7.5 mL pH值10.5的NaOH水溶液;反應釜密封后,恒溫油浴中加熱到140 ℃,磁力攪拌3 h。反應結束后,將反應釜在冷水中急速冷卻至室溫,經過濾分離,得到含有甲醇木質素的處理液。將該處理液經真空旋轉蒸發濃縮至5 mL,然后滴加到50 mL pH值2的鹽酸(體積分數0.3%)中,所得懸濁液在室溫下靜置過夜后過濾分離,將濾餅用去離子水洗至中性,冷凍干燥48 h,干燥后的濾餅,即為甲醇木質素。
1.4 木質素氫解制芳香化學品
將0.5 g的木質素、0.05 g Pd/C催化劑和10 mL甲醇/水(體積比1 ∶1)溶液加入到水熱釜中,密封后放入油浴中加熱至所需的反應溫度,反應一定時間后,將水熱釜迅速冷卻至室溫,然后過濾分離釜內混合物。濾餅用反應溶劑洗至無色,烘干后稱質量,計算固體殘渣質量。
濾液為木質素的氫解產物及溶劑。取1 mL濾液與等體積含有十二烷內標物的二氯甲烷混合,萃取,取下層萃取液用氣相色譜測定氫解產物中芳香族單體的含量。剩余濾液在35 ℃下真空干燥4 h,除去溶劑,所得油狀物質稱質量后采用氣相色譜分析單體含量。可溶物得率和芳香族單體產率計算公式如下:
式中:ys—可溶物得率,%;mr—固體殘渣質量,g;mL—起始木質素加入量,g;ya—芳香族單體產率,%;ma—芳香族單體質量,g。
1.5 分析與表征
1.5.1HPLC分析 毛竹水解渣及提取到的木質素的組分分析方法參考文獻[5]。準確稱量0.3 g絕干樣品,置于耐壓瓶中,加入3 mL 72%的硫酸,攪拌均勻,置于30 ℃恒溫水浴中反應1 h。反應得到的固體殘渣為Klason木質素及灰分,上清液為多糖水解產物和少量酸溶木質素。將固體殘渣在550 ℃馬弗爐中煅燒4 h,煅燒后得到灰分質量,除去灰分后的固體質量即為木質素質量。上清液用碳酸鈣中和后離心,然后采用高效液相色譜儀檢測上清液組成,流動相為純水,流速0.6 mL/min,柱溫85 ℃,檢測器溫度35 ℃,進樣量10 μL。
1.5.2GC-MS分析 采用GC-MS儀進行分析,FID為檢測器。以高純氮氣為載氣,正十二烷作為內標,柱箱的初始溫度為50 ℃,以10 ℃/min的升溫速率升溫至250 ℃并保持5 min。產物首先使用GC-MS分析,所得圖譜經過與NIST數據庫比對后進行定性。
1.5.3FT-IR分析 在FT-IR儀上進行分析。將樣品與溴化鉀按照質量比1 ∶100混合磨碎并制成透明薄片,以純溴化鉀壓片,在波數500~4000 cm-1,分辨率為4 cm-1的條件下進行分析。
1.5.4GPC分析 木質素及產品的相對分子質量測定采用凝膠色譜(GPC)法。過氧化氫木質素相對分子質量測定參考文獻[15]報道的方法,將5 mg樣品溶解在5 mL純水中,溶解后液體采用水系濾膜過濾,使用純水作為流動相,流速為0.5 mL/min,進樣量50 μL,檢測器為示差折光檢測器,以已知相對分子質量的葡聚糖為標準品進行標定。甲醇木質素及降解產物的相對分子質量測定參考文獻[16]報道的方法,將5 mg樣品溶解在5 mL色譜純四氫呋喃中,溶解后液體采用有機系濾膜過濾,四氫呋喃作為流動相,流速為1 mL/min,進樣量50 μL,檢測器為示差折光檢測器,以已知相對分子質量的聚苯乙烯樣品為標準品進行標定。
2 結果與討論
2.1 木質素的提取
毛竹水解渣以木質素和纖維素為主,含有少量的半纖維素。水解渣得率68.2%,水解液中木糖和阿拉伯糖的得率分別為原料總質量的20.5%和1.4%,由此計算得水解渣的纖維素、半纖維素和木質素質量分數分別為65.9%、4.9%和29.2%。以毛竹水解渣為原料,采用過氧化氫法提取木質素,木質素提取率為97.9%。醇析后得到的過氧化氫木質素占毛竹水解渣總質量的27.2%,其中木質素的質量分數為91.6%,其余為多糖組分。采用甲醇法提取木質素,木質素提取率為76.4%。酸析后得到的甲醇木質素占毛竹水解渣總質量的26.7%,其中木質素的質量分數為77.1%,葡聚糖質量分數22.3%,灰分質量分數0.6%。從木質素提取率和木質素產品純度分析,過氧化氫法優于甲醇法。
2.2 木質素氫解工藝條件探討
2.2.1過氧化氫木質素氫解產物分布 表1給出了不同反應溫度和反應時間條件下,過氧化氫木質素轉化得到的芳香族物質的分析結果。對過氧化氫木質素氫解轉化的產物做進一步分析,可以將其分為兩類:對位為脂肪族含氧取代的愈創木酚衍生物1~4(第I類)和對位為烷烴取代的愈創木酚衍生物及愈創木酚5~8(第II類)。
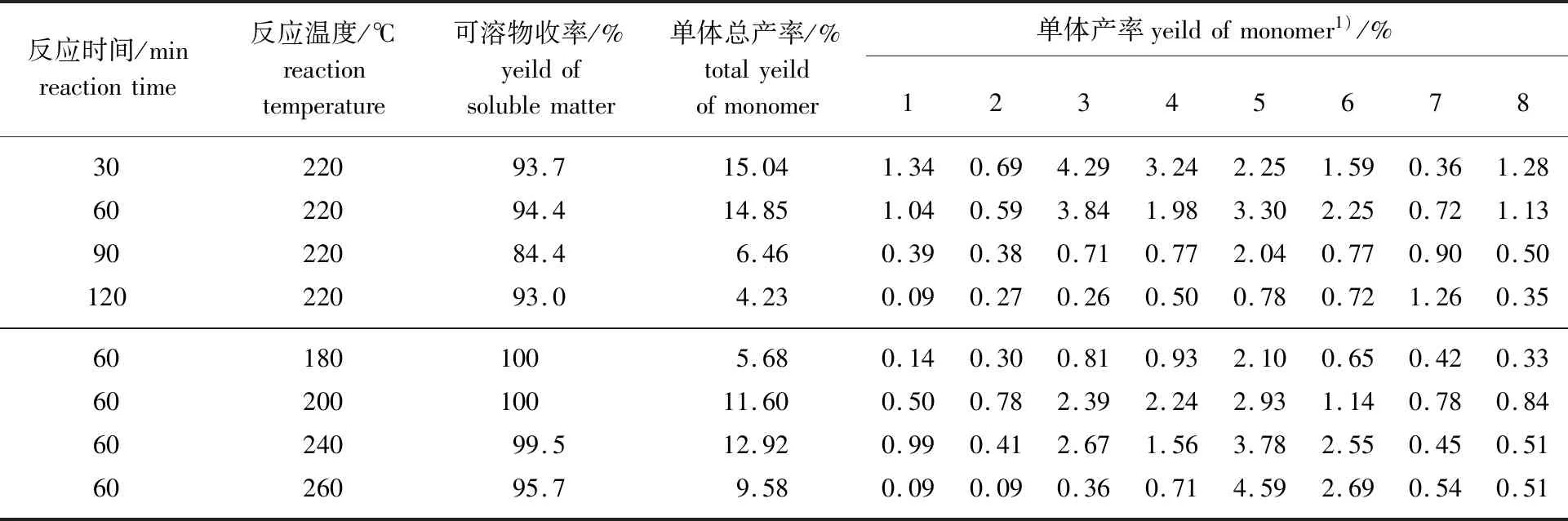
表1 反應條件對過氧化氫木質素氫解的影響Table 1 Effect of reaction conditions on hydrogenolysis of hydrogen peroxide lignin
表1還給出了這兩類產物收率隨反應溫度和反應時間變化的趨勢。可以看出,隨著氫解反應時間的延長,第I類產物的總收率先增加后降低,反應時間大于60 min后明顯降低。第I類產物的總收率從60 min時的7.44%降至120 min時的1.11%。這類產物總收率的降低是由每種產物收率降低的疊加導致的。其中,降低最明顯的物質為產物3,從3.84%降至0.26%。對于第II類物質,產物5和6在反應時間為60 min時產率最高。隨反應時間延長,產物7的收率逐漸升高,而產物8的收率在逐漸降低。隨著時間的延長,可溶物的收率呈現降低的趨勢,當反應時間大于90 min后降低最為明顯。不溶物部分可能含有未反應的木質素和木質素氫解產物二次反應生成的不溶性固體。此外,在220 ℃、反應時間為30~90 min時,木質素氫解產物二次反應很少,但反應時間大于90 min后會有明顯的增加。由此可知,氫解反應時間對于過氧化氫木質素的轉化及產物分布均有較大的影響,延長反應時間使芳香族單體物質的總收率降低,而其中一些物質的收率可能會出現極大值,在反應時間大于90 min后,芳香族單體物質的二次反應會明顯增加。
表1還給出了反應時間為60 min時,反應溫度對產物收率的影響。隨著反應溫度的提高,第I類產物的收率呈現先升高后降低的趨勢。第II類產物的總收率逐漸提高,其中產物5和6的收率隨著反應溫度升高而提高最為明顯,當反應溫度為260 ℃時,產物5和6的收率分別為4.59%和2.69%。產物7和8的收率則隨著溫度的提高呈現先升高后降低的趨勢。隨著溫度升高,木質素轉化的可溶物的收率在220 ℃時最高。這一現象可能意味著,溫度低于220 ℃時有少量的木質素沒有轉化,而溫度高于220 ℃ 時,氫解產物的二次反應會加劇。綜合上述的結果可以認為,以Pd/C作為催化劑,以甲醇/水為反應溶劑,氫解過氧化氫木質素時,反應溫度220 ℃,反應時間60 min,氫解產物得率最高。該反應條件將進一步用于甲醇木質素降解,考察其轉化效率。
2.2.2兩種木質素氫解產物對比 生物煉制過程,有機溶劑木質素(甲醇、乙醇或二氧六環木質素等)被認為是除磨木木質素外,最接近原生結構的木質素。在很多模型化合物轉化的研究中,大多會采用有機溶劑木質素做進一步驗證[17]。因此,本研究對比了相同反應條件(反應溫度220 ℃,反應時間60 min)下甲醇木質素的轉化情況。
甲醇木質素在相同反應條件下,轉化結果與過氧化氫木質素有明顯差別。甲醇木質素氫解轉化的芳香單體產物包括4-(4-羥基苯基)-2-丁酮0.95%,4-(4-羥基-3-甲氧基苯基)-2-丁酮2.00%,3-(4-羥基-3-甲氧基苯基)-1-丙醇1.13%和3-(4-羥基-3,5-二甲氧基苯基)-1-丙醇3.05%,單體總產率為7.13%,可溶物收率為96.8%。過氧化氫木質素的氫解產物以愈創木基型(G型)為主,酚羥基對位的脂肪族官能團的碳數為0~3;甲醇木質素的氫解產物包括愈創木基型(G型),紫丁香基型(S型)和對羥苯基型(H型)3種,酚羥基對位的脂肪族官能團碳的數目為3或4。除了產物種類的明顯差別外,兩種木質素轉化得到的芳香化學品的含量也存在較大差別。在220 ℃反應60 min 后,過氧化氫木質素氫解得到的芳香族物質總量為14.84%,而甲醇木質素氫解得到的芳香族物質總量僅為7.10%。過氧化氫木質素氫解得到的香草酮(3)、香草醛(4)分別為3.84%和1.98%,這兩種芳香化學品都是比較重要的香料或有機合成中間體。
2.3 木質素氫解產物分析
2.3.1相對分子質量分布 木質素是一種天然高分子物質,其氫解產物中除上述的芳香族單體物質外也含未降解的高分子物質。因此,也需要對這些高分子物質的性質進行分析,以便對于木質素及其降解產物的性質有較全面的了解。
圖1給出了過氧化氫木質素、甲醇木質素及其降解產物的GPC圖譜。過氧化氫木質素相對分子質量較大,其數均相對分子質量(Mn)為44 529,氫解后降至1 397,表明其發生了很明顯的降解;甲醇木質素的數均相對分子質量為1 581,氫解后降至1 183,降低并不明顯。
從分散系數(Mw/Mn)可以看出,過氧化氫木質素的分散系數為1.06,氫解后分散系數為1.46,說明氫解產生了更多的低相對分子質量產物,使相對分子質量分布范圍變廣。與之相反,甲醇木質素的分散系數在氫解前后分別為1.39和1.16,說明降解后甲醇木質素的相對分子質量分布范圍變得更為集中。兩種木質素相對分子質量分布的結果表明:過氧化氫木質素在氫解過程中比甲醇木質素更容易發生降解。
2.3.2FT-IR分析 圖2給出過氧化氫木質素、甲醇木質素及降解產物的FT-IR譜圖。
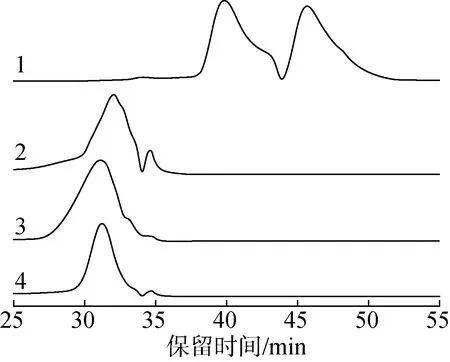

1.過氧化氫木質素hydrogen peroxide lignin;2.過氧化氫木質素降解產物degradation product of hydrogen peroxide lignin;3.甲醇木質素methanol lignin;4.甲醇木質素降解產物degradation product of methanol lignin
信號歸屬參考Wang等的研究[16]。從圖中可以看到,過氧化氫木質素降解后指紋區(1000~2000 cm-1)的紅外吸收信號數量增多,吸收峰位置發生了顯著的變化;甲醇木質素在降解后紅外吸收信號的降低相對較小,且吸收峰的位置沒有明顯的變化。在3403 cm-1處的寬吸收峰為O—H伸縮振動,對應木質素中脂肪族基團;2939 和1458cm-1處的吸收峰為甲基和亞甲基C—H伸縮振動;2841和1427 cm-1處的吸收峰為甲氧基中C—H伸縮和變形振動。
從圖2可看到,過氧化氫木質素在1458 cm-1處的吸收峰明顯強于甲醇木質素,該信號歸屬于甲基和亞甲基基團。此處的明顯差異表明,過氧化氫提取木質素會破壞苯環結構,發生開環,使苯環上共軛的碳氫轉變為非共軛的碳氫,從而提高脂肪族碳氫(如亞甲基)的相對含量,降低苯環信號的比例。

2.4 兩種木質素的結構差異分析
對于過氧化氫木質素和甲醇木質素結構的差異,可以結合過氧化氫與木質素的反應機理(圖3)進行解釋。堿性條件下,H2O2分解成過氧酸根離子(HOO-),使脂肪族α-C和γ-C分別轉變為過羥基和醛基,過羥基進一步脫氫氧根,變成氧負離子[18-19],此時木質素氧醚鍵斷裂的解離能較低,更容易發生解聚。此外,過氧化氫還有可能氧化苯環,使其轉變為苯醌甚至開環:苯環甲氧基在堿性條件下受到過氧根離子的攻擊,連接上一個氧負離子和一個過氧基,該結構會進一步轉化為一對鄰位苯醌。如果繼續有過氧根離子存在,鄰位苯醌之間的C—C就會發生斷裂,形成兩個帶有負電荷的酯鍵。
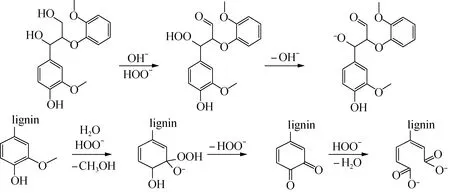
圖3 過氧化氫氧化木質素機理Fig.3 Mechanisms of delignification process by alkaline hydrogen peroxide oxidation
除了官能團被修飾外,氧化還能夠消除木質素分子內氫鍵,使大分子結構更加線性舒展。研究者利用密度泛函理論,結合半經驗公式,對8個苯環組成的木質素的分子構象進行計算,受分子內的氫鍵影響,分子骨架呈折疊狀態,寬度僅4.1 nm,任意兩個原子之間寬度小于1.5 nm[15]。破壞氫鍵后得到相對線性的大分子木質素,有利于非均相催化反應的進行。
FT-IR分析說明過氧化氫木質素轉化產品中芳香酸、酯和醛類多于甲醇木質素的轉化產物,解釋了氫解生成較多的第I類芳香族單體的原因。過氧化氫木質素反應前后指紋區信號的明顯變化,說明氫解可以使被過氧化氫氧化的木質素還原成芳香族化合物。
甲醇木質素在該體系中轉化為單體的含量相對于文獻報道是較低的[3,17]。這可能是由于反應過程中,針對氧醚鍵的定向轉化選擇性不高,采用的催化劑和溶劑效率不足導致的[4,20]。但是本研究對兩種木質素轉化研究,采用的催化劑和溶劑均為木質素轉化實驗常用的反應體系。過氧化氫法提取毛竹水解渣中的木質素轉化為芳香化學品的結果,證實其相比于甲醇法更具有優勢。
3 結 論
3.1分別采用過氧化氫法和甲醇法,從毛竹水解渣中提取獲得了兩種木質素,將木質素氫解轉化制芳香化學品,在反應溫度220 ℃,反應時間1 h的優化條件下,過氧化氫木質素可以全部轉化為可溶物,單體得率為14.85%;甲醇木質素的可溶物得率為96.8%,而單體得率僅為7.13%。
3.2對比了兩種木質素的氫解產物,過氧化氫木質素轉化產物以愈創木基型為主,酚羥基對位取代基團以羰基、醛基和烷烴取代基為主;FT-IR和GPC分析結果表明相比于甲醇木質素,過氧化氫木質素存在結構上的優勢,更容易通過催化氫解,轉化為芳香化學品。
猜你喜歡
中學生數理化·中考版(2022年10期)2022-11-10 09:37:42
中學生數理化·八年級物理人教版(2022年12期)2022-02-14 07:08:42
中學生數理化·八年級物理人教版(2021年12期)2021-12-31 03:23:08
中學生數理化·中考版(2020年10期)2020-11-27 01:59:48
中國生殖健康(2019年2期)2019-08-23 08:12:08
石油化工建設(2018年6期)2018-04-22 03:16:54
產品可靠性報告(2017年7期)2017-09-05 09:49:12
中學生數理化·八年級物理人教版(2017年12期)2017-04-18 12:59:38
汽車觀察(2016年3期)2016-02-28 13:16:26
民生周刊(2014年7期)2014-03-28 01:30:54
