Chromosomal assembly of the Antarctic toothfish(Dissostichus mawsoni) genome using third-generation DNA sequencing and Hi-C technology
2021-03-01 06:52:30SeungJaeLee,Jeong-HoonKim,EunaJo等
Zoological Research 2021年1期
DEAR EDITOR,
The Antarctic toothfish,Dissostichus mawsoni,belongs to the Nototheniidae family and is distributed in sub-zero temperatures below S60° latitude in the Southern Ocean.Therefore,it is an attractive model species to study the stenothermal cold-adapted character state.In this study,we successfully generated highly contiguous genome sequences ofD.mawsoni,which contained 1 062 scaffolds with a N50 length of 36.98 Mb and longest scaffold length of 46.82 Mb.Repetitive elements accounted for 40.87% of the genome.We also inferred 32 914 protein-coding genes usingin silicogene prediction and transcriptome sequencing and detected splicing variants using Isoform-Sequencing (Iso-Seq),which will be invaluable resource for further exploration of the adaptation mechanisms of Antarctic toothfish.This new high-quality reference genome ofD.mawsoniprovides a fundamental resource for a deeper understanding of cold adaptation and conservation of species.
Many unique fish live in the Southern Ocean surrounding Antarctica within the coldest waters on Earth.It has been isolated by the Antarctic circumpolar current (Eastman,2005;Livermore et al.,2005) where sea temperatures range around the ice point (–1.9 °C) for most of the year.Antarctic fish,which include cold-adapted teleosts,are dominated by a single lineage belonging to the Perciformes suborder Notothenioidei.This suborder consists of eight families and>100 species and accounts for~90% of total fish biomass in the Antarctic Ocean (Eastman & De Vries,1981).From a common ancestor,a variety of closely related species with distinct ecological status as well as size,shape,and color have emerged in the Southern Ocean.Therefore,genomic research is essential to understand the environmental adaptation and evolution of these fish.
The Antarctic toothfish,Dissostichus mawsoni,belongs to the family Nototheniidae of the order Perciformes and is native to the Southern Ocean.It is distributed below S60° latitude and is the largest of all Antarctic fish (2.0 m in length and 140 kg in mass) (Eastman & De Vries,1982).Its stenothermal cold-adapted state makes the species an attractive model for evolutionary and genomic studies among Antarctic fish.The Antarctic toothfish is an economically important fishery species,with a commercial catch in Subarea 88.1 of 2 680 tons in 2018 (Maschette et al.,2019)
Recently,de novoassembly of the Antarctic toothfish genome and extensive transcriptomic characterization using short-read Illumina data have been reported (Chen et al.,2019),though the genome was fragmented into many scaffolds due to sequencing by synthesis technology.The development of third-generation single-molecule sequencing technology has enabled the production of long-read sequences and the discovery of the features of previously unavailable DNA regions.Here,we report on a re-assembled whole-genome ofD.mawsoniusing long-read sequencing and Hi-C technology,which should help provide comprehensive insight into its adaptive mechanisms.
Antarctic toothfish (length~50 cm,sex not determined)were collected using a vertical setline in the eastern Ross Sea(Subarea 88.1),Antarctica (http://www.fao.org/fishery/area/Area88/en),during the austral summer season (December 2018).Specimens were killed for tissue sampling and then rapidly frozen for further analysis.All sample collection and experimental protocols were in compliance with the laws regarding activities and environmental protection in Antarctic and were approved by the Minister of Foreign Affairs and Trade of the Republic of Korea.
To obtain sufficient high-quality DNA molecules for the PacBio Sequel platform (Pacific Biosciences,USA),oneD.mawsonifish was dissected and muscle tissue was used for DNA extraction using the phenol/chloroform extraction method.DNA quality was checked using a fragment analyzer system (Agilent Technologies,USA) and Qubit 2.0 fluorometer(Invitrogen,Life Technologies,USA).The single-molecule real-time (SMRT)bell library was sequenced using eight SMRT cells (Pacific Biosciences,SequelTMSMRT Cell 1M v2) with a Sequel Sequencing Kit 2.1 (Pacific Biosciences,USA) and 1×600 min movies were captured for each SMRT cell using the Sequel sequencing platform (Menlo Park,USA).The average coverage of the SMRT sequences was~81-fold(Supplementary Table S1).
Muscle tissue from the same sample was used to construct a Hi-C chromatin contact map for chromosome-level assembly.Tissue fixation,chromatin isolation,and library construction were performed according to the manufacturer’s instructions (Dovetail Genomics,USA) (Belton et al.,2012).After checking the insert size,concentration,and effective concentration of the constructed libraries,the final libraries were sequenced using the Illumina NovaSeq 6 000 platform(San Diego,USA) with a 150-bp paired-end strategy.A total of 874 million raw reads were generated from the Hi-C libraries(Supplementary Table S1) and were mapped to the polishedD.mawsonicontigs using HiC-Pro (v2.8.0) with default parameters.
For transcriptome sequencing,we prepared 1 μg of pooled total RNA from the muscle and skin ofD.mawsoni.Using a SMARTer PCR cDNA Synthesis Kit (Clontech,USA),RNA was synthesized to cDNA. The SMRTbell library was constructed using the SMRTbellTMTemplate Prep Kit 1.0-SPv3.The SMRTbell library was sequenced using SMRT cells(Pacific Biosciences,SequelTMSMRT Cell 1M v2) and the Sequel Sequencing Kit 2.1.For each SMRT cell,1×600 min movies were captured with the use of the Sequel sequencing platform; the pre-extension time was 240 min (Supplementary Table S1).The Iso-Seq sequencing data were analyzed using SMRT Link (v6.0.0).
Forde novogenome assembly, the FALCON-Unzip assembler (v0.4,Falcon,RRID:SCR 016089) was used(Chin et al.,2013) with parameters of length_cutoff=12 000 and length_cutoff_pr=10 000 and with filtered subreads from SMRT Link (v4.0.0) (minimum subread length=50). To improve the quality of the genome assembly,the FALCONUnzip assembler was polished using the Arrow algorithm with unaligned BAM files as raw data.
A draftD.mawsonigenome was previously generated using Illumina short-read sequencing (Chen et al.,2019).However,since several gaps prevailed in the draft genome set and there was no information about the linkage group,it was difficult to compare the structure of the Antarctic toothfish genome at the chromosomal scale.To improve this genome resource,longread SMRT sequencing from Pacific Biosciences and Hi-C scaffolding were implemented.First,we performedde novoassembly of the long PacBio sequence reads using the FALCON-Unzip tool and obtained a genome assembly with a size of 924.75 Mb,an N50 contig size of 3.23 Mb,and longest contig size of 24.49 Mb (Supplementary Table S2).To construct the reference genome at the chromosome level,we constructed a Hi-C library and anchored the scaffolds into chromosomes after quality control using the HiC-Pro,Juicer(v1.5) (Durand et al.,2016) and 3D-DNA (v170123) pipeline(Dudchenko et al.,2017) based on the draft genome assembly(Figure 1A).The assembled genome was 926.3 Mb (GC content:41.57%) in length with a scaffold N50 of 36.98 Mb and longest scaffold of 46.82 Mb.In total,there were 1 062 scaffolds in theD.mawsonigenome assembly,with 24 chromosome-scale scaffolds occupying 91.3% of the assembly (Supplementary Tables S2,S3 and Figure S1).
Benchmarking Universal Single-Copy Orthologs (BUSCO)v3.0 (Sim?o et al.,2015) (RRID:SCR 015008) was used along with the actinopterygii odb9 database to assess the completeness of the newD.mawsonigenome assembly.Among 4 584 BUSCO groups searched,4 197 and 194 BUSCO core genes were completely and partially identified,respectively,contributing to a total of 95.7% BUSCO genes in theD.mawsonigenome (Supplementary Table S4).
The diploid chromosome number (2n) ofD.mawsoniis 48(Ghigliotti et al.,2007).Comparison of its chromosome-scale assemblies with those of theGasterosteus aculeatusgenome(2n=42) showed a highly similar synteny (Figure 1B).However,each of three chromosomes (from Groups 1,4,and 7) ofG.aculeatusmatched with two chromosomes inD.mawsoni(scaffolds 23 and 35,scaffolds 13 and 22,scaffolds 11 and 24,respectively) (Supplementary Figure S2).
Ade novorepeat library was constructed using RepeatModeler (v1.0.3) (Bao & Eddy,2002),which included RECON (RRID:SCR 006345) and RepeatScout v1.0.5(RRID:SCR 014653) (Price et al.,2005) software with default parameters.The Tandem Repeats Finder (Benson,1999)program was used to predict the consensus sequences and classification information for each repeat.We analyzed the repetitive sequences in theD.mawsonigenome,including those in the tandem repeats and transposable elements (TEs).TEs play an important role in shaping genome architecture and are a source of regulatory mutations in evolution.A difficulty arises in representing TEs in genome assemblies based on short Illumina sequence reads.Therefore,our longread sequences greatly improved both the length and quantity of the TE repeats in theD.mawsonigenome assembly compared to the published short-read assembly.TheD.mawsonigenome contained 40.87% of repeat sequences,including 36.27% (336 Mb) of TEs such as long terminal repeats (LTRs,4.21%),short interspersed nuclear elements(SINES,0.50%),long interspersed nuclear elements (LINEs,(5.35%),and DNA transposons (15.51%) (Supplementary Table S5 and Figure S3).Divergence of TEs was examined using RepeatMasker software, where Kimura distances(Kimura, 1980) estimated for aligned TEs(calcDivergenceFromAlign.pl) were used to draw repeat landscapes (createRepeateLandscape.pl).TheD.mawsonigenome had a higher number of recent TE insertions (Kimura divergenceK-values≤5) that were strongly shaped by DNA transposons (Supplementary Figure S4).BecauseK-values calculated for TEs can reflect age and transposition history(Chalopin et al.,2015),we concluded that there have been recent transposable element bursts in the Antarctic toothfish.
Genome annotation was conducted using MAKER v2.28(RRID:SCR_005309) (Holt & Yandell,2011),which is a portable and easily configurable genome annotation pipeline.Subsequently,repeat masked genomes were used forab initiogene prediction with SNAP v2006–07-28 (SNAP,RRID:SCR 002127) (Korf, 2004) and Augustus (Augustus: Gene Prediction,RRID:SCR_008417) software.MAKER was initially run in the est2genome mode based on the Iso-Seq data,including 57 406 full-length transcripts.Additionally,protein evidence was obtained from the genomes of 19 teleosts,including three Antarctic fish (Supplementary Table S6).Exonerate software,which provides integrated information for the SNAP program,was used to polish MAKER alignments.MAKER was then used to select and revise the final gene model considering all available information.Other non-coding RNAs in the Antarctic toothfish assembly were identified using Infernal (v1.1) (Nawrocki & Eddy,2013) and covariance models (CMs) from the Rfam database v12.1 (Rfam,RRID:SCR 007891) (Griffiths-Jones et al.,2005).Putative tRNA genes were identified using tRNAscan-SE v1.3.1(tRNAscan-SE,RRID:SCR 010835) (Lowe & Eddy,1997),which uses a CM that scores candidates based on their sequences and predicted secondary structures.
The predicted genes were aligned to the NCBI nonredundant protein (nr) (Benson et al.,1999),SwissProt(RRID:SCR_002380) (Boeckmann et al.,2003),TrEMBL(RRID:SCR_002380) (Boeckmann et al., 2003), KOG(EuKaryotic Orthologous Groups) (Tatusov et al.,2001),and KEGG (Kyoto Encyclopedia of Genes and Genomes,RRID:SCR_001120) (Kanehisa & Goto,2000) databases using BLAST v2.2.31 (Altschul et al.,1990) with a maximum e-value of 1e-5.Gene Ontology (GO) (RRID:SCR_002811)terms (Dimmer et al.,2012) were assigned to the genes using the Blast2Go v4.0 pipeline (RRID:SCR_005828) (Conesa et al.,2005).
A total of 32 914 protein-coding genes in theD.mawsonigenome were annotated using a combination ofab initiogene prediction,homology searching,and transcript mapping.The coding sequence comprised 51.2 Mb (exons 55.2 Mb) with an average of 9.7 exons per gene (Supplementary Table S7).Consequently,a total of 20 202 genes were annotated in >1 database (Supplementary Table S7).A total of 24 920,19 205,and 14 474 genes were annotated in the GO,KOG,and KEGG databases, respectively, and the functional classifications of these genes are presented in Supplementary Figures S5–S7.
We identified orthologous gene clusters using the OrthoMCL (Li et al.,2003) pipeline,which applied the Markov Clustering Algorithm (MCL) with default options in all steps for the genome sequences of the 20 species (Supplementary Table S6).It was critical for analysis to include representative species of diverse phylogenetic clades and the 20 species were selected among those with well-annotated and wellassembled genomes.
Phylogenetic tree construction was performed based on single-copy orthologous genes.The sequences of proteincoding genes were aligned using a Probabilistic Alignment Kit(PRANK) (L?ytynoja & Goldman,2005) with the codon alignment option. The maximum-likelihood method was applied to construct a phylogenetic tree using RAxML with 1 000 bootstraps,and divergence times were calibrated with TimeTree (median estimates of pairwise divergence time forD.rerioandG.morhua:230.4 million years ago) (Hedges et al.,2006).
Ortholog gene families from each species were identified:7 731 orthologous gene families were commonly identified among the 20 teleosts, including 434 (1 431 genes)paralogous gene families that wereD.mawsoni-specific(Supplementary Table S8).The phylogenetic tree ofD.mawsoniand the 19 teleost species was constructed using 1 422 single-copy orthologs (Figure 1C).Among the 20 fish species,D.mawsoniand three other Antarctic fish were clustered together on the branch of a non-Antarctic fish,G.aculeatus,with a divergence time of around 105 million years ago.Furthermore,D.mawsonidiverged approximately 28 million years ago from the Antarctic fishChaenocephalus aceratus.Analysis of gene gain-and-loss among genomes enables the reconciliation of a species tree with the gene tree for each family.Here,D.mawsonihad 659 (including 2 114 genes) significantly expanded and 116 (including 136 genes)significantly contracted gene families (Figures 1C,D).The vast majority of the expanded biological pathways belonged to two functional categories:(i) involved in nervous system functions (neuron projection development, GO:0 031175;neuron development, GO:0 048666; cell morphogenesis involved in neuron differentiation,GO:0 048667; generation of neurons,GO:0 048699; neuron projection morphogenesis,GO:0 048812; axon development, GO:0061564) and (ii)cellular component morphogenesis (cell projection organization, GO:0 030030; cell part morphogenesis,GO:0 032990; cell projection morphogenesis,GO:0048858).In the molecular function category,peptidase regulator activity(endopeptidase regulator activity,GO:0 061135; peptidase inhibitor activity,GO:0 030414; endopeptidase inhibitor activity,GO:0004866),and signaling receptor binding (endopeptidase inhibitor activity,GO:0004866) were the major expanded pathways (Supplementary Tables S9,S10).In addition,14 055 orthologous gene families containing 16 162 genes inD.mawsoniwere commonly identified in the four Antarctic fish.Moreover,621 gene families wereD.mawsonispeciesspecific paralogs (Figure 1E) involved in DNA metabolic processes (DNA biosynthetic process,GO:0 071897; DNA integration,GO:0 015074; RNA-dependent DNA biosynthetic process,GO:0006278) (Supplementary Table S11).
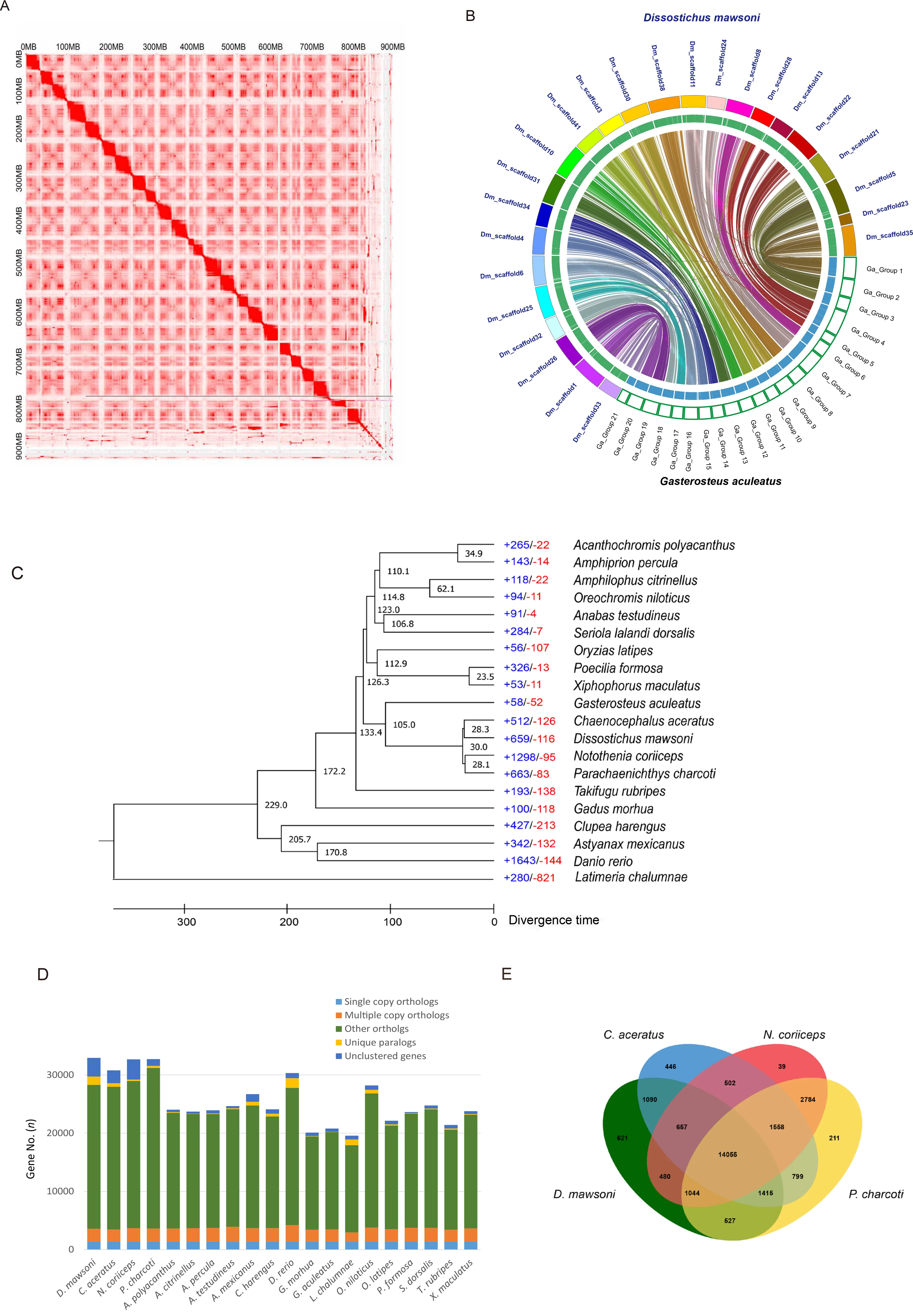
Figure 1 Genome analysis of D.mawsoni
Splicing variants were analyzed using SQANTI2 (Tardaguila et al.,2018) with the Iso-Seq data as full-length transcript sequences.The Iso-Seq data were aligned to the assembled genome using Minimap2 (Li,2018) and the collapsed highquality isoforms were aligned to unique isoforms using the Cupcake ToFU pipeline (Tseng,2017).SQANTI2 extracted various types of splicing variants using the collapsed isoforms and the gene prediction information of the assembled genome.After excluding novel,mono exonic,and antisense transcripts,genes were selected based on the ascending order of the number of isoforms.Enrichment analysis of genes of splicing variants was performed using the Blast2GO v4.0 pipeline (RRID:SCR_005828) (Conesa et al.,2005) with FDR<0.5.
Iso-Seq data analysis identified 31 480 unique isoforms in 14 565 unique genes.Most novel genes were located in the intergenic region (Supplementary Tables S12, S13).Functional annotation using enrichment analysis by Fisher’s Exact Test for genes with more than 10 splicing variants(Supplementary Table S14) identified genes related to development,anatomical structure development,functional annotations (system development,GO:0 048731; animal organ development, GO:0 048513;tissue development,GO:0 009888; cell development, GO:0 048468; embryo development,GO:0 009790; muscle structure development,GO:0 061061; epithelium development,GO:0 060429; and circulatory system development, GO:0072359), and organization related function (cytoskeleton organization,GO:0 007010;protein-containing complex subunit organization,GO:0 043933; actin cytoskeleton organization,GO:0 030036; supramolecular fiber organization,GO:0 097435;and organelle organization,GO:0006996) (Supplementary Table S15).
In the current study, we presented a high-quality chromosome-level genome assembly of the Antarctic toothfish,D.mawsoni,using PacBio Sequel sequencing and Hi-C chromatin contact maps.TheD.mawsonigenome assembly (926 Mb) included 24 chromosomes that accounted for 91% (840 Mb) of all genome sequences.TheD.mawsonigenome contained 32 914 protein-coding genes and 434 paralogousD.mawsoni-specific gene families among 20 teleost fish and 621 paralogousD.mawsoni-specific gene families among the four Antarctic teleost fish. This chromosome-length genome assembly will not only provide insights into the molecular and ecological adaptations of Antarctic fish to extreme environments but will also facilitate exploration of genomic adaptations to a wide range of evolutionary, ecological, metabolic, developmental, and biochemical features of Antarctic fish.
DATA AVAILABILITY
TheDissostichus mawsonigenome project was deposited in NCBI under BioProject No.PRJNA574770 and the Whole-Genome Shotgun project was deposited at DDBJ/ENA/GenBank under accession No.JAAKFY000000000.The version described in this paper is JAAKFY010000000.The genome browser, assembly, and annotation data are accessible on http://genome.kusglab.org/.
SUPPLEMENTARY DATA
Supplementary data to this article can be found online.
COMPETING INTERESTS
The authors declare that they have no competing interests.
AUTHORS’ CONTRIBUTIONS
H.P.and H.-W.K.conceived the study.S.J.L.,E.J.,S.-G.C.,S.C.E.C.,J.K.,and H.P.performed genome sequencing,assembly, and annotation. E.J. and S.J.L. performed experiments.S.J.L.,J.-H.K,H.-W.K.,and H.P.mainly wrote the manuscript.All authors contributed to writing and editing the manuscript as well as collating the supplementary information and creating the figures.All authors read and approved the final version of the manuscript.
ACKNOWLEDGEMENTS
We would like to thank Sunwoo Corporation for providing the Antarctic toothfish samples.

- Zoological Research的其它文章
- The forty-year journey of Zoological Research:advancing with the times
- Geographical range evolution of the genus Polypedates (Anura:Rhacophoridae) from the Oligocene to present
- Molecular and morphological evidence for a new species of the genus Typhlomys (Rodentia:Platacanthomyidae)
- Pitfalls of barcodes in the study of worldwide SARSCoV-2 variation and phylodynamics
- Contribution to the taxonomy of the genus Lycodon H.Boie in Fitzinger,1827 (Reptilia:Squamata:Colubridae) in China,with description of two new species and resurrection and elevation of Dinodon septentrionale chapaense Angel,Bourret,1933
- Dynamic evolution of transposable elements,demographic history,and gene content of paleognathous birds