基于細菌16S rRNA 基因擴增子測序數據的系統發生樹圖制作方法
2021-01-21 06:45:48閻星羽廉振穎成麗娟刁玉濤王家林
醫學信息 2021年1期
丁 可,閻星羽,廉振穎,成麗娟,刁玉濤,王家林
(1.山東第一醫科大學,山東 泰安 271016;2.山東第一醫科大學基礎醫學院/基礎醫學研究所,山東 濟南 250062;3.山東中醫藥大學第二附屬醫院,山東 濟南 250001;4.山東第一醫科大學附屬腫瘤醫院,山東 濟南 250117)
細菌微生物16S rRNA 基因擴增子二代測序數據分析的后續任務之一是通過系統樹形圖(dendrogram/phylogenetic tree)展示微生物種群之間在進化或種系發生上的相互關系情況,dendrogram/phylogenetic tree 圖分3 種類型,分別為進化分支圖(cladogram),僅有拓撲結構,不能從數量上說明各分支間進化距離的大小;系統發生(發育)圖(phylogram),各分枝長度表示堿基替換數,因而能從數量上說明各分支間進化距離的大小;時序圖(chronogram)則用各分枝長度表示進化時間,其中系統發生圖(phylogram)無疑是微生物分子遺傳學研究最常用到的圖形化方法[1]。目前系統發生圖制作軟件生成的系統發生樹文件為Newick 和Nexus 格式,并且大多數系統發生樹查看軟件也主要兼容上述2 種文件格式,但Newick 和Nexus 格式的樹文件只包含物種間的遺傳距離和進化拓撲關系,結構比較單一。而R語言中的“ggtree”包對ape、ggplot2 等繪圖包的功能進行了優化,“ggtree”除了能支持眾多的樹文件格式,其繪圖方法更豐富,繪制的圖形更準確和美觀,最大的優點是能通過各種附加信息對樹形圖進行多方面的注釋[2]。因此,本研究構建了微生物基因組擴增子二代測序數據的系統發生樹形圖的制作流程,通過QIIME 命令代碼對測序數據進行預處理,聚類生成OTUs 表數據,對OTUs 表格數據進行篩選和轉換,并用QIIME 分析管道(pipeline)的相關命令生成Newick 格式的系統發生樹文件,以此文件為輸入,用R 語言“ggtree”包的函數和代碼繪制包含更多研究信息的不同類型的系統發生樹圖,現將結果報道如下。
1 材料與方法
1.1 數據來源 分析數據來自文獻所用的原始測序數據[3],基于人體微生物在不同部位并隨時間推移的變化,通過QIIME 分析方法,只選取其中的部分數據,即每天分別從2 個人的舌部、左手手掌、右手手掌和腸道共取得34 個標本,在Illumina HiSeq 2000 平臺上進行微生物16s rDNA 擴增子測序。此外,從“https://data.qiime2.org/2017.6/tutorials/moving-pictures/emp -single -end -sequences/barcodes.fastq.gz”網站獲得經過整理后的單向測序的數據。
1.2 軟件系統 通過QIIME 完成系統發育樹構建和可視化的工作流程,并使用是R 語言中的“ggtree”包(3.6.0 版)進行樹文件的可視化和注釋。
1.3 分析流程 構建可視化系統發生樹的工作流程,關鍵步驟是形成BIOM(Biological Observation Matrix)格式的OTUs 表。QIIME 系統中有多個Python腳本用于生成OTUs 表,按聚類算法分為使用從頭聚類方法(de novo)和使用參考數據庫聚類方法。在此工作流程中,本研究使用的是基于Greengene 參考數據庫的封閉參考(Closed reference)聚類分析方法生成OTUs 表數據進行分析的,具體步驟如下:①生成OTUs 表文件(otu_table.biom):目的是依據上一步生成的序列文件seqs.fna,通過與最新的Green-Genes 數據庫比對進行聚類分析,其主要輸出是OTUs 表格(out_table.biom),表格中的內容為記錄每個OTU 在每個樣品(微生物群落標本)中被觀察到的次數;②OTUs 的過濾:通過filter_otus_from_otu_table.py 命令代碼篩選OTUs,只保留相對含量>1‰(或其它比例)的OTUs;③OTUs 表文件格式轉換:目的是將上一步生成的biom 格式的OTUs 表文件轉換為純文本(csv、tsv 或txt 格式)文件,利于下一步數據處理;④選取代表序列:由于每個OTU 中的序列不完全相同,因此需要通過pick_rep_set.py 腳本選取一條代表性序列作為該OTU 的序列,用于后續分析;⑤物種分級注釋:用assign_taxonomy.py 腳本命令對每個OTU 代表性序列進行物種分類信息的注釋,可以認為每個OTU 近似為一個物種,反之一個分類到“種(species)”或“屬(genus)”水平的物種類別可以對應一個到多個OTU;⑥代表序列的比對:QIIME 系統的align_seqs.py 腳本提供3 種序列比對方法,即Py-NAST、MUSCLE 和INFERNAL,本研究中使用QIIME 系統默認的PyNAST 方法,它基于NAST 算法,將輸入序列與提供的參考序列數據比對,在數據庫中找到最高匹配的序列;且MUSCLE 不需要提供參考序列,可用于真菌轉錄間隔區(internal transcribed spacer,ITS)測序比對分析;而INFERNAL 利用RNA結構和序列相似性進行比對,與PyNAST 一樣需要比對數據庫;⑦篩選比對序列:由于上述align_seqs.py 腳本通過將長度200~400 bp 的目的序列和16S rRNA 基因的全序列比對,因此,生成的代表性序列包含空缺(gaps),為了保留代表性序列中的有用信息以構建系統發育樹,需要通過filter_alignment.py 腳本對上述代表性序列進行篩選,去除堿基空缺等無用信息;⑧建樹:運用python make_phylogeny.py 腳本構建系統發育樹;⑨樹文件的圖形化:通過FigTree軟件和R 語言“ggtree”包對Newick 格式的系統發育樹文件進行圖形化處理。系統發育樹文件生成與可視化的數據處理工作流程見圖1。
2 結果
本結果來自對34 個取樣標本16S rRNA 基因測序數據的分析,34 個標本分別來自人體的不同部位和不同取樣時間,為方便起見,本分析流程沒有考慮采樣時間,僅將不同取樣部位作為分組因素進行分析。首選通過QIIME-1.9.1 系統的pick_closed_reference_otus.py 腳本生成OTUs 表和系統發生樹文件,然后根據物種分類信息,將物種名稱標記到“樹葉”上,構建優化的物種聚類樹文件,該樹文件作為“ggtree”包的輸入文件,再結合研究設計的分組信息繪制不同類型的系統發生樹圖。
2.1 樣本信息和OTUs 聚類結果 本示例數據來自人體的腸道(gut)、左側手掌(left palm)、右側手掌(right palm)、和舌部(tongue),為了分析方便,省略了取樣時間、抗生素使用情況等其它分組信息。通過封閉參考(closed reference)聚類法將有效測序序列按照≥97%的相似性歸為一個OTU。采用RDP 算法與Greengene 16S rRNA 數據庫比對,并將各OTU注釋到所屬的分類單元。使用Greengene 數據庫可以將大多數OTU 序列注釋到“屬”水平,少部分OTU可以鑒定到“種”水平。通過上述方法將示例數據中的共177882 條序列聚類成4403 個OTU,在“門”和“屬”兩個水平上對同類的OTU 進行分類鑒定,分別歸屬于24 個門,615 個屬,有關樣本的人體部位分組情況、OTU 聚類和種屬鑒定的具體情況見表1。OTUs 聚類及分類鑒定結果與同時生成的系統發生樹文件,可以作為后續統計分析,構建系統發生樹圖的基礎。
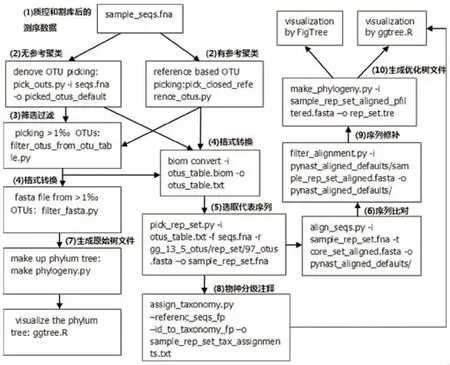
圖1 系統發育樹文件生成與可視化的數據處理工作流程
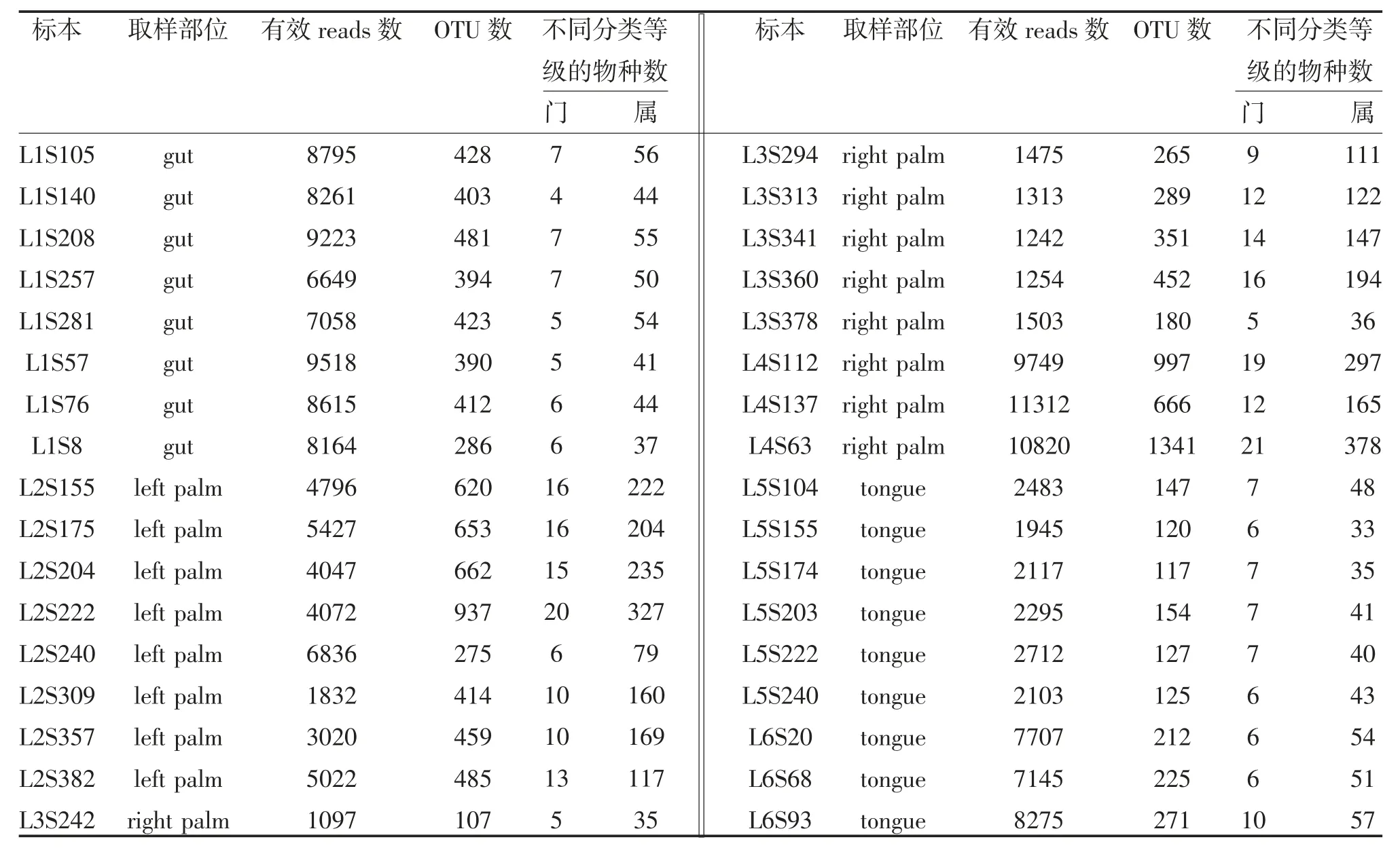
表1 樣本測序序列與OTU 聚類情況統計表
2.2 各OTU 的物種歸類 人體不同部位的微生物構成存在顯著差異,通過QIIME 系統的種群多樣性分析命令集core_diversity_analyses.py 可全面分析不同取樣部位之間物種的兩類多樣性指數Alpha 多樣性指數(α-diversity)與Beta 多樣性指數(β-diversity)的差異,更重要的是可以發現人體特定部位的菌群相對含量(豐度)并發現其優勢菌群。在“門”和“屬”水平對菌群分類結果顯示,具體菌群分類與豐度信息可以在多樣性分析輸出目錄(“taxa_summary_plots”)中查看,該命令集還會給出不同物種的豐度在樣本組(人體部位)之間差異的統計學假設檢驗結果(“group_significance_SampleType.txt”),其中擬桿菌屬(bacteroides) 在腸道中的相對含量為60.48%,遠高于其它身體部位(P<0.05)而鏈球菌屬(streptococcus)只存在與手掌部和舌部,在腸道未檢出(P<0.05),見圖2。
2.3 QIIME 系統生成的原始系統發生樹 FigTree 軟件打開原始系統生成樹文件,FigTree 的運行需要JAVA 語言支持,需根據軟件提示安裝相應的JAVA運行環境,依次點擊file--open--rep_set3.tre 輸入原始樹文件rep_set3.tre,運行后生成rectangular、polar 和radial 3 種類型的系統發生樹圖,結果顯示其只有物種間的進化距離關系,但“樹葉”只能用OTU的編碼表示,rectangular 形狀的樹形圖見圖3。
2.4 通過QIIME 和R 語言“ggtree”包優化系統發生樹
2.4.1 優化樹形 為了使樹文件中包含更少的樹枝以增加圖形的可分辨性,將篩選OTUs 的豐度閾值定為5%。在R 語言運行環境下調用ggtree 包讀取篩選后的Newick 格式的樹文件rep_set4.tre 繪圖,定義樹形圖的顏色、線條的形狀和樹形圖布局顯示物種間進化距離及比例尺、標注內部節點和樹枝末端、顯示OTUs 編號。根據以上條件繪制的樹形圖見圖4。
2.4.2 向樹圖中添加物種分類信息 通過QIIME 的assign_ assign_taxonomy.py 腳本將篩選后的OTU 代表序列進行物種注釋,即每個OTU 對應一個物種名稱,將物種注釋文件轉換為csv 格式并命名為sample_rep_set4_tax_assignments.csv,需要給文件中的每一列命名,其中第一列“taxa”即為系統發生樹文件中的OTU 編號,“taxonomy”列為精確到“種”水平的物種分類信息,最后通過“%<+%”操作符將“taxonomy”列中與OTU 對應的物種分類信息添加到系統發生樹中。運行R 代碼生成的系統發生樹圖見圖5。

圖2 示例樣品菌群分類與相對豐度累積條圖
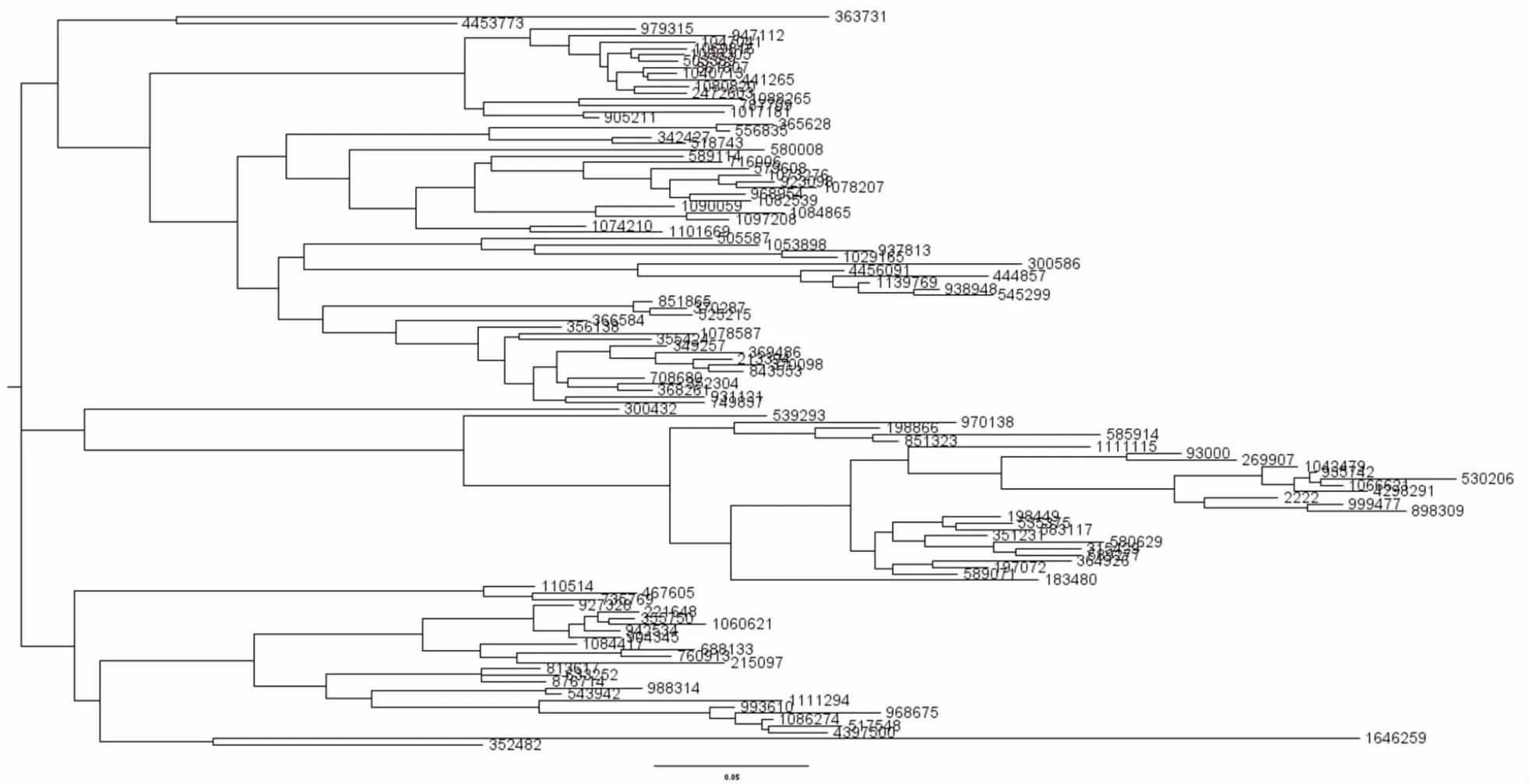
圖3 FigTree 軟件查看rectangular 布局的原始系統發生樹圖
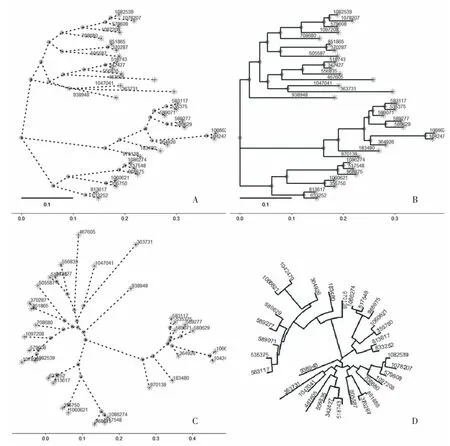
圖4 “ggtree”包生成的沒有物種注釋的系統發生樹

圖5 ggtree 包繪制的包含物種分類信息注釋的系統發生矩形樹圖
2.4.3 系統發生和物種豐度聯合圖 通過QIIME 的convert_biom.py 腳本將篩選后的biom 格式的OTUs表文件轉換為純文本格式并命名為table7.from_biom_w_consensuslineage.txt,文件中第一列taxa 即為≥5%豐度的OTU 編號,其它列為每個OTU在不同標本中的豐度(相對含量)值。運行R 語言代碼生成的系統發生樹與OTU 豐度熱圖見圖6,可以比較方便的查看不同OTU 間進化親緣關系的遠近以及OTU 數量在每個標本中的分布情況。同時,還可以按研究設計的標本分組情況將標本進行合并后作圖,對于本示例數據,可以按取樣部位將標本合并為腸道(gut)、手掌(palm)和舌部(tongue)來源共3 個組,或者將所有標本合并為1 個組后,重新繪制系統發生和OTU 在不同組間豐度分布的組合圖,運行代碼生成的系統發生樹與OTU 豐度分布熱圖,結果見圖7,其中圖7B 所示的聯合圖系將所有標本合并后繪制系統發生樹、點圖和條形圖的聯合圖,點的位置和條圖的長短均表示相應的OTU 的豐度值。
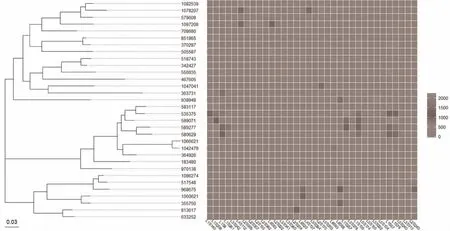
圖6 系統發生樹和OTU 在不同標本中的豐度分布聯合圖
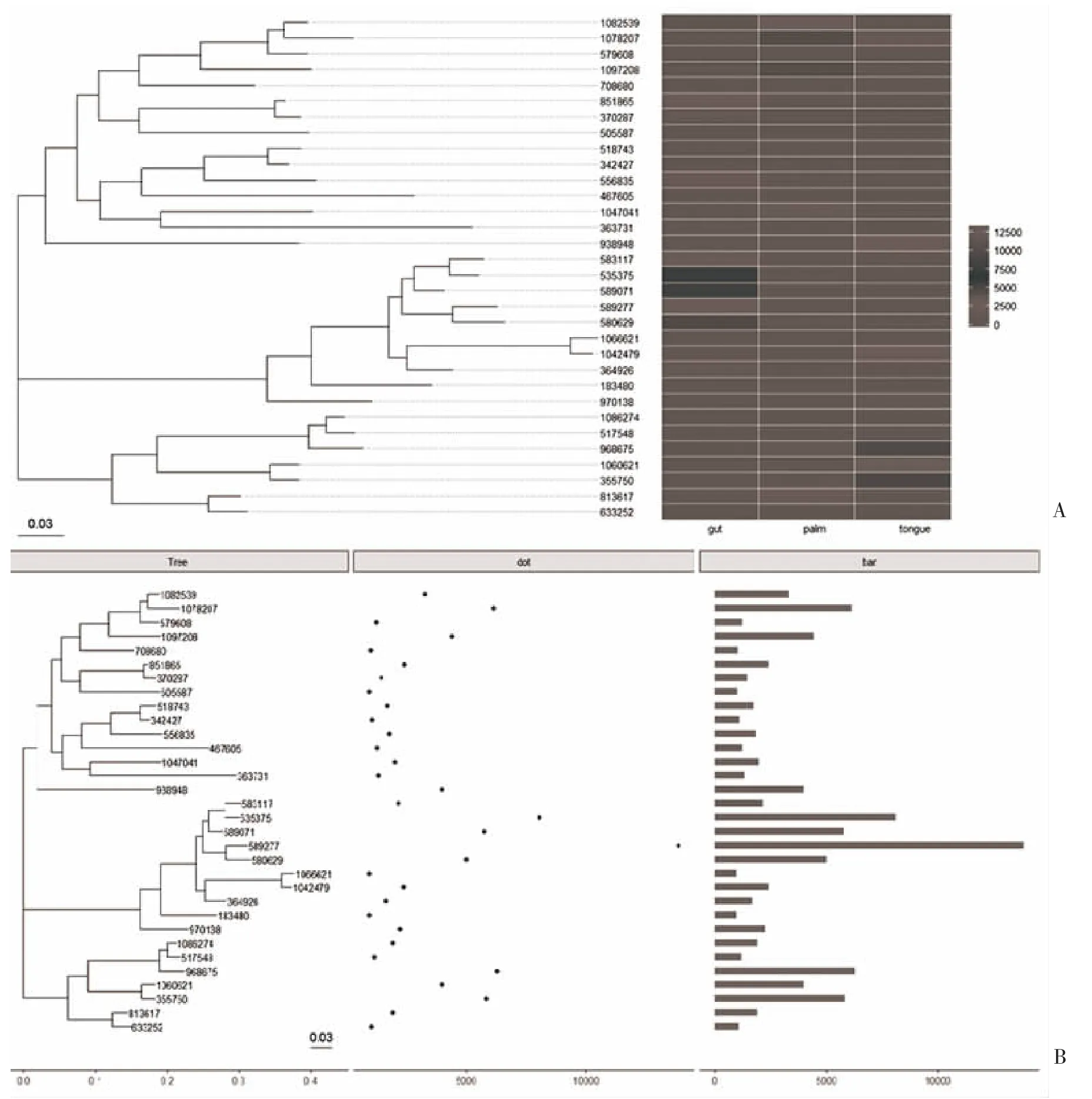
圖7 系統發生樹和OTU 在不同標本組中的豐度分布聯合圖
3 討論
細菌16S rRNA 基因是編碼原核生物核糖體小亞基的基因,長度約為1542 bp,在細菌進化過程中的突變率相對較小,并且其分子大小適中,是細菌系統分類學研究中最常用和最有用的分子標志[4]。通過對該基因的V3~V4 單(或雙)可變區域進行PCR 擴增和二代測序(NGS),根據測序數據預測標本中微生物群落的種屬信息和種屬的相對構成比,并進一步研究標本中微生物群落的種屬多樣性、微生物與環境因子的相互關系,以及微生物功能基因組與宿主和環境因子之間的相互依存關系,這些均是目前微生物學、環境科學和醫學研究中的重要課題[5-8]。
用于細菌16S rRNA 基因二代測序數據分析的常用軟件主要有 Mothur、QIIME、Uparse、RDP、VAMPS、MEGAN 等,其中Mothur 和QIIME 是最常用的分析工具。QIIME 和R 語言相結合可以解決幾乎全部的微生物二代測序數據的整理、統計分析和分析結果的可視化處理。QIIME 的主要用途是運用經過整理后的測序數據,通過聚類分析生成可操作的分類單元(OTUs)數據表明,OTUs 表中的數據為微生物群落的種群分類和數量組成信息。以OTUs表數據作為輸入,通過R 語言中的相關程序包可對OTUs 表中的數據進行圖形化展示。R 語言作為開源的,面向對象的交互式語言,除了能進行常規的數據處理和統計學分析,近年來有眾多學者針對分子生物學實驗數據處理的要求開發了大量的數據處理工具,并將這些工具連同部分實驗數據以包的形式放置到R 語言環境當中,使用者通過相應代碼(命令)調用這些包完成相應的分析任務。在上述分析任務中,通過系統發生樹圖展示微生物種群之間在種系發生上的相互關系是微生物組數據分析的主要內容。目前多數生物信息學軟件生成的微生物組系統樹文件只包含最基本的物種間的拓撲關系,結構比較單一。因此本研究設計了基于QIIME 系統和R 語言“ggtree”包的構建系統發生樹圖操作流程,通過該操作流程,研究者可以向系統發生樹圖中添加實驗設計信息,繪制內容更加豐富的系統發生樹圖,滿足論文發表和研究報告的生成等科研需求。
本工作流程整合了專業分析細菌16S rRNA 基因測序數據的QIIME 系統和用于數據分析和圖形化展示的R 語言系統。該方法的特點在于分析過程全部在Windows 系統下進行,其中QIIME 的工作界面雖然是基于linux 系統的,但可以通過VirtualBox在windows 系統中虛擬出一個Linux 系統。并且本流程是基于代碼的操作,與其他商業化程序相比具有很高的應用靈活性。更重要的是,工作流程是從原始的測序數據出發的,而不是從由Windows 和菜單用戶界面的軟件生成的樹文件開始,因此可以繪制多種類型的系統發生樹形圖。使用本流程繪制的圖形更準確和美觀,最大的優點是能通過各種附加信息對樹形圖進行多方面的注釋,還可以針對系統發生與微生物豐度聯合作圖。
猜你喜歡
課堂內外·初中版(科學少年)(2025年1期)2025-02-28 00:00:00
課堂內外·初中版(科學少年)(2025年2期)2025-02-28 00:00:00
英語世界(2023年10期)2023-11-17 09:18:18
工業設計(2022年8期)2022-09-09 07:43:20
軍民兩用技術與產品(2021年10期)2021-03-16 06:05:30
北京測繪(2020年12期)2020-12-29 01:33:58
裝備制造技術(2019年12期)2019-12-25 03:06:46
科學大眾(中學)(2019年3期)2019-05-17 10:04:30
中國洗滌用品工業(2019年4期)2019-05-11 09:27:34
汽車觀察(2018年10期)2018-11-06 07:05:26
