ICP-MS法檢測干貨類調味料中10種有害元素殘留
2021-01-18 09:08:08崔曉娜郭禮強彭曉蓓
食品工業 2020年12期
崔曉娜,郭禮強,彭曉蓓
1. 山東畜牧獸醫職業學院(濰坊 261061);2. 濰坊海關(濰坊 261041)
鉛、砷、鎘、汞、銅、鉻、錫、鎳、鋅、錳[1]是自然界中主要的有害元素,可以借助水源和食物鏈被人體吸收,誘發多種癌癥,有些還會傳給下一代[2]。目前對有害元素的研究主要在土壤和農作物方面,關于調味料的報道較少。調味料一般在貧瘠的土地上種植,更容易遭到有害元素的污染。
干貨類調味料有害元素檢測方法主要有石墨爐原子吸收光譜法[3]、火焰原子吸收光譜法[4]、原子熒光光譜法[5-7]、電感耦合等離子體原子發射光譜法[8]和電感耦合等離子體質譜法[9-12]。石墨爐和火焰原子吸收光譜法通量低,精度和重現性較差;原子熒光光譜法對復雜基體的樣品測定比較困難;原子發射光譜法檢測時容易出現譜線重疊干擾、耐鹽量差、痕量分析靈敏度不及電感耦合等離子體質譜法。根據GB 2762—2017《食品安全國家標準 食品中污染物限量》的限量要求,此次試驗以多種有害元素為研究對象,結合微波預處理技術,采用電感耦合等離子體質譜(ICPMS)法建立了10種有害元素的定量檢測方法,該方法具有較高的靈敏度和實用性,可為政府執法部門和食品生產加工企業提供必要的技術支持。
1 試驗部分
1.1 儀器與試劑
電感耦合等離子體質譜儀(7900,美國安捷倫公司);制水機(Milli-Q,德國密理博公司);微波消解儀(WX-8000,上海屹峣公司);高速組織搗碎機(DS-1,上海儀昕科學儀器公司);電子天平(MLT,瑞士梅特勒公司);研磨儀(德國萊馳公司);移液器(1~5 mL,德國赫施曼公司);硝酸(65%,德國默克公司);高純氬氣(99.99%以上,山東昌霖氣體公司)。
1.2 標準溶液
混合多元素標準儲備液:22種元素混合標準溶液,含有銀、鋁、鉛、砷、鋇、鉍、鈣、鎘、汞、銅、鉻、鈷、錫、鉀、鋰、鎳、鋅、鐵、錳、鎂、鈉、硒,其中鈣、鉀、鋅、鐵、鈉的質量濃度為1 000.0 μg/mL,其他元素的質量濃度為10.0 μg/mL(美國安捷倫公司);汞標準儲備液(1 000.0 μg/mL,青島捷世康公司);
ICP-MS調諧液:10 μg/mL,用3%硝酸溶解,鈰、鈷、鋰、鈧、鉈、釔(山東省冶金科學研究院標準樣品研究所);ICP多元素內標液:1.0 μg/mL,鋰、鈧、鍺、釔、銦、鋱、鉍(上海甄準生物科技公司)。
混合多元素標準工作液:移取適量混合多元素標準儲備液,用5%硝酸系列稀釋(現用現配)。汞標準工作液:移取適量汞標準儲備溶液,用2%鹽酸系列稀釋(現用現配)。
1.3 微波消解處理條件(見表1)
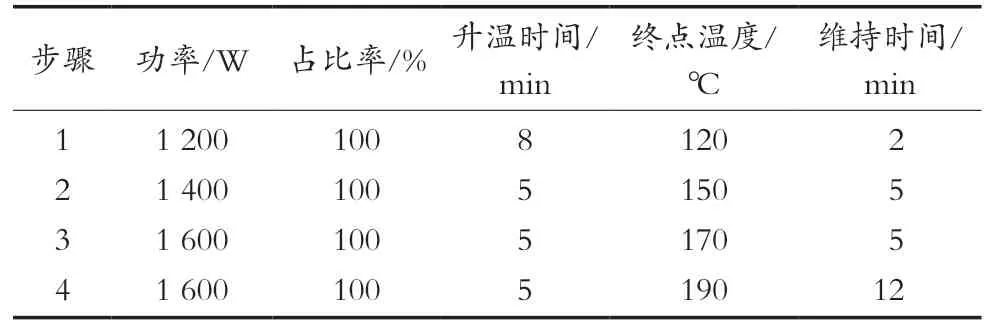
表1 微波設定程序
1.4 質譜條件
霧化器,石英同心霧化器;霧化室,石英雙通道,控溫2.0±0.1 ℃;循環水流速1.0 mL/min;蠕動泵流速0.1 mL/min;炬管,石英材質,中心直徑2.5 mm;儀器功率1 550 W;采樣深度8 mm;反射功率<10 W;載氣流速900 mL/min;在線內標1.0 μg/mL。
1.5 質譜調諧液
1.0 μg/mL靈敏度范圍:鋰>3 000,釔>8 000,鈰>4 000。氧化物:氧化鈰/鈰<1%;鈷>7 000,鈧>6 000,鉈>8 000。雙電荷:Ce2+/Ce+<3%。
1.6 樣品處理
將干貨調味料樣品先混合均勻再用四分法縮分,然后隨機取樣,盛裝到潔凈的合適容器中,用搗碎機將樣品打成粉末,準確稱取0.2 g于聚四氟乙烯消解高壓罐中,用移液器加入8 mL硝酸,蓋好塞子旋緊,靜置4 h后放入保護套中微波消解,消解程序見1.3,結束后消解罐中液體全部轉移至潔凈塑料瓶,用去離子水定重至50 g,待測定。
2 結果與討論
2.1 同位素的選擇
在電感耦合等離子體質譜檢測中,碰撞池采用He模式,通過調查待測目標元素的原子量,選擇各元素最穩定含量最高的同位素和原子量接近的內標物來消除離子干擾、樣品基體效應和信號漂移。同時,由于自然界中有害元素天然存在,檢測時造成數據偏移,試驗通過在線加入混合內標的方式來提高待測元素的準確度和穩定性。用內標物鉍元素校正鉛和汞元素,用內標物鍺來校正鉻、鎳、銅、鋅、錳、砷元素,用銦內標物來校正鎘和錫元素。
2.2 靈敏度的優化
八角、桂皮等樣品比較堅硬,含有大量的纖維素、揮發油、色素和礦物質,尤其是鉛、鎘殘留較多,樣品不均會造成檢測結果失真,單一元素含量過高,超過標準曲線的線性范圍會抑制目標元素的離子化,導致檢測數據偏低。因此,樣品一定要粉碎均勻,在確保質譜檢出限的基礎上,減少樣品稱樣量,將樣品稀釋50倍,能較好地解決基質干擾和元素抑制問題。通過對2012—2018年濰坊地區進出口干貨類調味品中元素數據分析,確定1.6的步驟參數,基本滿足多種樣品的檢測需求,適合干貨類調味品復雜基質中有害元素的檢測。若遇到污染嚴重超出線性范圍的樣品,需要用5%硝酸或2%鹽酸(汞元素檢測)進一步稀釋樣品,確保樣品上質譜檢測濃度在目標元素標準品線性范圍之內。
2.3 方法的線性關系和檢出限
混合標準溶液系列用5%硝酸配制(汞標準品用2%鹽酸配制),以各有害元素的質量濃度為橫坐標,以各目標元素與其參照內標標準品的色譜響應值相對比率為縱坐標,制作各個有害元素的標準曲線。選取干貨類調味品中較難處理的代表性樣品桂皮,按1.6小節進行處理,對桂皮空白基質樣品溶液連續12次平行檢測,根據GB/T 27404—2008測定低限的要求計算樣品檢測結果的平均值和標準偏差,以10倍的標準偏差作為定量限(LOQ),部分參數見表2。從表2看出,10種目標分析物標準品在0.1~100.0 μg/L質量濃度范圍內線性關系良好,相關系數均大于0.999,LOQs在0.001~0.050 mg/kg之間。
2.4 方法的回收率及精密度
為了檢驗方法的有效性,按1.6小節進行處理,分別在桂皮空白樣品中添加各個目標元素方法定量限、2倍定量限和10倍定量限的標準溶液,每個添加濃度做6個平行樣,經質譜檢測后計算各個目標物的回收率和精密度(相對標準偏差)。由表3可知,在3個添加濃度下10個目標物的平均回收率范圍在87.0%~115.7%之間,相對標準偏差(RSD)在2.0%~9.2%之間,滿足GB/T 27404—2008附錄F對精密度的檢測要求。
2.5 實際樣品測定
對市場上抽取的20份干貨類調味料(八角、花椒、桂皮和肉桂各5份)樣品中的有害元素殘留進行了分析檢測,從一份肉桂樣品中檢測到鉛,含量為1.53 mg/kg,而國家標準GB 2762—2017中調味品的限量要求是≤1.0 mg/kg,已經超標,建議政府相關部門進一步加強對調味料中有害元素的監管。
3 結論
此次試驗建立了ICP-MS快速測定干貨類調味料中10種有害元素殘留的方法,能夠實現對10種有害元素同時定性定量分析。該方法操作簡便,靈敏度高,實用性強,可以滿足對干貨類調味料中多種有害元素殘留的檢測需求。
猜你喜歡
城市道橋與防洪(2022年4期)2022-07-01 06:04:12
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2020年12期)2021-01-18 06:57:46
中學生數理化·七年級數學人教版(2020年12期)2021-01-18 06:57:46
當代陜西(2019年8期)2019-05-09 02:22:48
動漫星空(興趣百科)(2019年3期)2019-03-07 07:23:10
家庭影院技術(2018年4期)2018-05-09 07:07:52
海峽科技與產業(2016年3期)2016-05-17 04:32:12
