β2-AR別構(gòu)拮抗劑Cmpd 15關(guān)鍵中間體(S)-9-芴基甲氧羰基-4-甲酰胺基苯丙氨酸的合成
2021-01-04 13:18:32錢明成
合成化學(xué) 2020年12期
趙 帥, 芮 雪, 錢明成, 陳 新
(常州大學(xué) 藥學(xué)院,江蘇 常州 213164)
G蛋白偶聯(lián)受體(G protein-coupled receptors, GPCRs)是體內(nèi)最龐大的蛋白質(zhì)超家族,目前已經(jīng)有2000多種不同的GPCRs被報道[1-2]。G蛋白偶聯(lián)受體通過與細(xì)胞周圍環(huán)境中的配體結(jié)合從而激活細(xì)胞內(nèi)的一系列信號通路,引起細(xì)胞狀態(tài)的改變,參與了人體內(nèi)幾乎全部生理病理和藥理過程[3]。其功能與多種疾病有關(guān),如心血管類疾病、神經(jīng)系統(tǒng)疾病、炎癥疾病、代謝性疾病、癌癥等。目前,超過40%的現(xiàn)代藥物直接或間接靶向GPCRs[4]。
β腎上腺素受體(β-AR)是研究最深入的GPCRs之一[5-6]。其中,β2-AR在支氣管平滑肌、心血管和肺部都有廣泛表達(dá),在調(diào)節(jié)心血管系統(tǒng)各項(xiàng)活動中扮演著至關(guān)重要的作用。目前市售β2-AR激動劑和拮抗劑較多,如沙丁胺醇(salbutamol)、特布他林(terbutaline)、沙美特羅(salmeterol)、福莫特羅(formoterol)和普萘洛爾(propranolol)等[7]。值得注意的是,上述β2-AR藥物皆為正構(gòu)配體,由于正構(gòu)配體的高度保守性和受體基因序列的高度同源性,這些正構(gòu)配體存在受體亞型選擇性差、神經(jīng)系統(tǒng)不良反應(yīng)和禁忌癥較多等問題[8]。與之相比,在空間和拓?fù)浣Y(jié)構(gòu)上,與正構(gòu)結(jié)合位點(diǎn)不同的其他區(qū)域相結(jié)合的別構(gòu)配體,因具有更高的受體亞型選擇性而成為藥物研發(fā)的熱點(diǎn)[9-14]。目前,已報道的β2-AR別構(gòu)調(diào)節(jié)劑有別構(gòu)激動劑Cmpd6FA[15]和別構(gòu)拮抗劑Cmpd15[16]。其中,Cmpd15的結(jié)構(gòu)于2017年由本課題組與美國合作者共同報道(Chart 1)[17]。如圖所示,Cmpd15可以由3個片段A~C偶聯(lián)得到。其中片段A廉價易得,片段C的合成已由本課題組報道[18-19]。然而片段B對甲酰基取代的L-苯丙氨酸十分昂貴(7200元/g,安耐吉化學(xué)),不利于后續(xù)進(jìn)行的Cmpd15衍生物的合成研究。
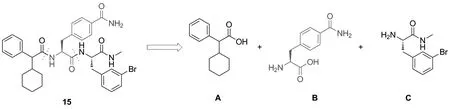
Chart 1
本文從二苯亞胺甘氨酸叔丁酯(2)和對甲酰胺芐溴(3)出發(fā),經(jīng)過不對稱烷基化、水解、脫保護(hù)基和酰胺化等多步反應(yīng)成功合成了Fmoc保護(hù)的片段B,即化合物12。通過在其羧酸端引入長鏈的聚乙二醇羧酸酯結(jié)構(gòu),成功測得了其ee值(97%)(Scheme 2),產(chǎn)物結(jié)構(gòu)經(jīng)1H NMR,13C NMR和HR-MS(ESI)確證。
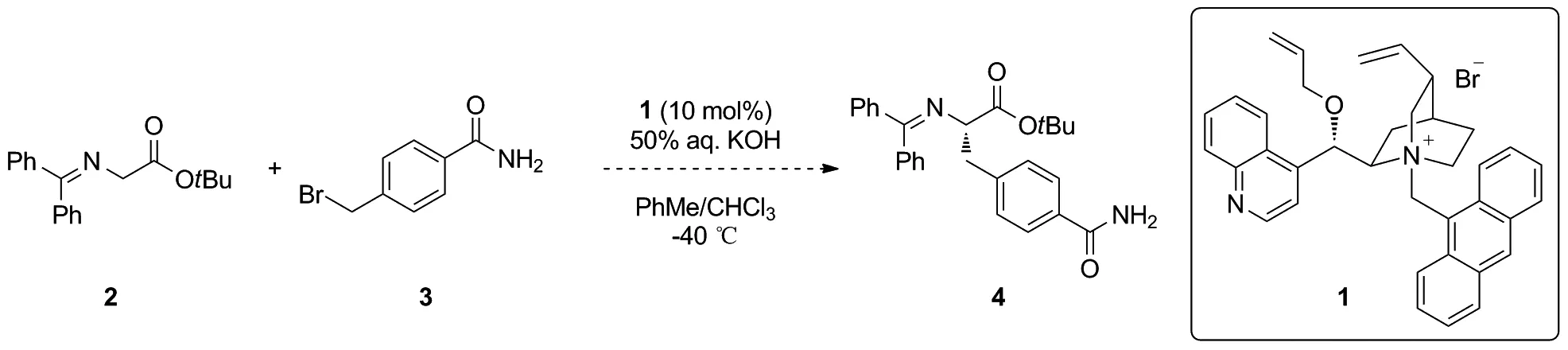
Scheme 1
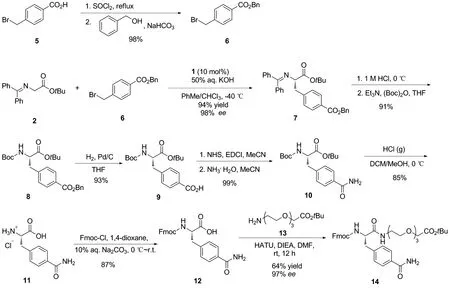
Scheme 2
1 實(shí)驗(yàn)部分
1.1 儀器與試劑
Autopol IV型旋光儀;Bruker 300/400 MHz型核磁共振儀(TMS為內(nèi)標(biāo));Thermo Orbitrap Elite型質(zhì)譜儀;Agilent 1260型高效液相色譜儀。
化合物1[20]、2[19]、3[21]和13[22]按文獻(xiàn)方法合成;其余所用試劑均為分析純。
1.2 合成
(1)4-溴甲基苯甲酸芐酯(6)的合成
在250 mL單口燒瓶中,加入4-溴甲基苯甲酸5(6.0 g, 27.9 mmol)和氯化亞砜(18 mL),回流反應(yīng)3 h。冷卻至室溫,濃縮除去過量氯化亞砜,將苯甲醇(24 mL)和NaHCO3(7.0 g, 83.7 mmol)加入濃縮液中,攪拌5 h。抽濾,濾液濃縮后經(jīng)硅膠柱層析[洗脫劑:V(石油醚)/V(乙酸乙酯)=100/1]純化得白色固體6(8.3 g,收率98%);1H NMR(CDCl3, 300 MHz)δ: 8.04(d,J=8.4 Hz, 2H), 7.32~7.45(m, 7H), 5.36(s, 2H), 4.47(s, 2H);13C NMR(CDCl3, 75 MHz)δ: 165.9, 142.9, 136.0, 130.3, 130.1, 129.1, 128.7, 128.4, 128.3, 66.9, 32.3。
(2)(S)-4-(3-(叔丁氧基)-2-((二苯基亞甲基)氨基)-3-氧亞基)苯甲酸芐酯(7)的合成
(3)(S)-4-(3-(叔丁氧基)-2-((叔丁氧基羰基)氨基)-3-氧亞基)苯甲酸芐酯(8)的合成
將(S)-4-(3-(叔丁氧基)-2-((二苯基亞甲基)氨基)-3-氧亞基)苯甲酸芐酯7(1.0 g, 1.93 mmol)溶于四氫呋喃(17 mL)中,降溫至0 ℃,滴加1 M HCl溶液(17 mL),滴畢,于0 ℃反應(yīng)1 h。加入飽和碳酸氫鈉溶液(10 mL)淬滅反應(yīng),用10% Na2CO3溶液調(diào)節(jié)pH至7~9,減壓濃縮,殘余物加入水(40 mL),用乙酸乙酯(3×40 mL)萃取,合并有機(jī)相,依次用飽和食鹽水洗滌,無水硫酸鈉干燥,減壓濃縮,粗產(chǎn)物溶于7 mL無水四氫呋喃中,于0 ℃滴加三乙胺(403 μL, 2.9 mmol)和二碳酸二叔丁酯(532 μL, 2.3 mmol),滴畢,反應(yīng)1 h。用水(40 mL)淬滅反應(yīng),加入乙酸乙酯(3×40 mL)萃取,合并有機(jī)相,依次用飽和食鹽水洗滌,無水硫酸鈉干燥,濃縮,粗產(chǎn)物經(jīng)硅膠柱層析[洗脫劑:V(石油醚)/V(乙酸乙酯)=20/1]純化得白色固體8(800.4 mg, 91%);1H NMR(CDCl3, 400 MHz)δ: 8.00(d,J=8.1 Hz, 2H), 7.32~7.45(m, 5H), 7.25(d,J=7.2 Hz, 2H), 5.35(s, 2H), 5.04(d,J=4 Hz, 1H), 4.45~4.50(m, 1H), 3.06~3.17(m, 2H), 1.41(d,J=7.2 Hz, 18H);13C NMR(CDCl3, 100 MHz)δ: 170.7, 166.4, 155.1, 142.2, 136.1, 129.8, 129.7, 128.8, 128.7, 128.3, 128.3, 82.5, 79.9, 66.8, 54.7, 38.6, 28.4, 28.1。
(4)(S)-4-(3-(叔丁氧基)-2-((叔丁氧基羰基)氨基)-3-氧亞基)苯甲酸(9)的合成
將化合物8(250.0 mg, 0.6 mmol)溶于甲醇(3 mL)中,加入鈀碳(37.5 mg, 15%),通入氫氣,反應(yīng)至終點(diǎn)。用硅藻土抽濾,濾餅濃縮得白色固體9(187.2 mg, 93%);1H NMR(DMSO-d6, 400 MHz)δ: 12.87(s, 1H), 7.85(d,J=8.0 Hz, 2H), 7.34~7.38(m, 2H), 7.24(d,J=8 Hz, 1H), 3.96~4.09(m, 1H), 3.01(dd,J=13.7 Hz, 5.6 Hz, 1H), 2.88~2.94(m, 1H), 1.29~1.34(m, 18H);13C NMR(DMSO-d6, 100 MHz)δ: 171.0, 167.3, 155.4, 143.1, 129.4, 129.2, 129.0, 80.5, 78.2, 55.6, 36.5, 28.1, 27.6。
(5)(S)-2-(((叔丁氧羰基)氨基)-3-(4-氨基甲酰基苯基)丙酸叔丁酯(10)的合成
氮?dú)獗Wo(hù)下,將9(150.0 mg, 0.4 mmol),N-羥基丁二酰亞胺NHS(61.2 mg, 0.5 mmol)和EDCI(103.3 mg, 0.5 mmol)溶于乙腈(5 mL)中,攪拌下反應(yīng)12 h。濃縮,殘余物加入水(10 mL),用二氯甲烷(3×10 mL)萃取,合并有機(jī)相,用無水硫酸鈉干燥,濃縮,殘余物用乙腈溶解,于0 ℃滴加氨水(1 mL),滴畢,攪拌1 h。濃縮,殘余物加入水(10 mL),用二氯甲烷(3×10 mL)萃取,合并有機(jī)相,依次用飽和食鹽水洗滌,無水硫酸鈉干燥,減壓濃縮,殘余物經(jīng)硅膠柱層析[洗脫劑:V(二氯甲烷)/V(甲醇)=20/1]純化得白色固體10(148.4 mg, 99%);1H NMR(DMSO-d6, 400 MHz)δ: 7.9(s, 1H), 7.79(d,J=8.0 Hz, 2H), 7.30(d,J=7.8 Hz, 2H), 7.20(d,J=8.0 Hz, 1H), 4.01~4.07(m, 1H), 2.99(dd,J=13.7 Hz, 5.4 Hz, 1H), 2.86~2.91(m, 1H), 1.30~1.35(m, 18H);13C NMR(DMSO-d6, 100 MHz)δ: 171.1, 167.7, 155.4, 141.2, 132.4, 129.0, 127.4, 80.5, 78.2, 55.7, 36.3, 28.1, 27.6。
(6)(S)-4-甲酰胺基苯丙氨酸鹽酸鹽(11)的合成
將10(138.0 mg, 0.4 mmol)溶于二氯甲烷(2 mL)和甲醇(2 mL)中,通入氯化氫氣體,反應(yīng)至終點(diǎn)(TLC監(jiān)測)。減壓濃縮,殘余物加入少量乙酸乙酯,抽濾,濾餅用乙酸乙酯洗滌得白色固體11(79.1 mg, 85%);1H NMR(DMSO-d6, 400 MHz)δ: 8.57(s, 3H), 7.89(d,J=8.1 Hz, 2H), 7.42(d,J=8.1 Hz, 2H), 4.19(s, 1H), 3.23(dd,J1=21.1 Hz,J2=14.8 Hz, 2H);13C NMR(DMSO-d6, 100 MHz)δ: 170.2, 140.3, 167.2, 129.8, 129.7, 129.5, 52.9, 35.5。
(7)(S)-9-芴基甲氧羰基-4-甲酰胺基苯丙氨酸(12)的合成
在25 mL的單口燒瓶中,加入化合物11(90.0 mg, 0.4 mmol),1,4-二氧六環(huán)和10%碳酸鈉溶液,于0 ℃緩慢滴加Fmoc-Cl(95.2 mg, 0.4 mmol)的1,4-二氧六環(huán)(1 mL)溶液,滴畢,攪拌下于0 ℃反應(yīng)4 h;升溫至室溫,反應(yīng)16 h。用3 N鹽酸溶液酸化反應(yīng)體系至pH=1,加入水(20 mL),用乙酸乙酯(3×20 mL)萃取,合并有機(jī)相,依次用飽和食鹽水洗滌,無水硫酸鈉干燥,減壓蒸餾,白色混合物加入少量乙酸乙酯,抽濾得白色粉末狀固體12(138.3 mg,產(chǎn)率87%);1H NMR(DMSO-d6, 400 MHz)δ: 7.86(t,J=8.5 Hz, 4H), 7.77(d,J=8.5 Hz, 1H), 7.62(t,J=6.6 Hz, 2H), 7.38~7.42(m, 4H), 7.25~7.42(m, 2H), 4.14~4.24(m, 4H), 3.16(dd,J=13.7 Hz, 4.0 Hz, 1H), 2.91~2.97(m, 1H);13C NMR(DMSO-d6, 100 MHz)δ: 170.8, 169.8, 169.5, 155.9, 143.9, 141.4, 141.2, 132.0, 129.7, 127.9, 127.8, 127.2, 125.2, 120.1, 82.1, 70.7, 70.5, 70.4, 70.2, 69.8, 68.9, 67.0, 56.1, 47.2, 39.4, 39.1, 28.2; HR-MS(ESI)m/z: Calcd for C25H22N2O5{[M+Na]+}453.1421, found 453.1424。
(8)(S)-5-(4-氨基甲酰基芐基)-1-(9-芴基甲酯)-3,6-二氧亞基-2,10,13,16-四氧亞基-4,7-二氮雜十八烷-18-酸酯(14)的合成
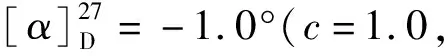
為了實(shí)現(xiàn)片段B簡潔高效的不對稱合成,首先嘗試使用對甲酰胺芐溴3與二苯亞胺甘氨酸叔丁酯2直接發(fā)生不對稱烷基化反應(yīng)構(gòu)建片段B。但是遺憾的是,反應(yīng)體系非常復(fù)雜,且產(chǎn)物很少(Scheme 1)。可能原因之一是化合物3在有機(jī)溶劑中的溶解性非常差。因此將對甲酰胺芐溴3中的酰胺替換為酯基后,與二苯亞胺甘氨酸叔丁酯2發(fā)生不對稱烷基化反應(yīng)。經(jīng)過一系列篩選,最終選定了對甲酸芐酯取代的芐溴6作為反應(yīng)底物。其中芐基可以通過催化氫化反應(yīng)轉(zhuǎn)化為相應(yīng)的羧酸,之后通過氨基化反應(yīng)得到片段B中的甲酰胺取代基。
從商業(yè)可得的對甲酸芐溴5出發(fā),經(jīng)過兩步反應(yīng)合成了所需的芐溴底物6。接下來芐溴6和二苯亞胺甘氨酸叔丁酯2在手性相轉(zhuǎn)移催化劑1的作用下,發(fā)生不對稱烷基化反應(yīng),以94%的產(chǎn)率和98%的ee得到了化合物7[20,23]。再經(jīng)過亞胺水解和Boc保護(hù)得到化合物8。其中水解反應(yīng)需要在0 ℃下進(jìn)行,防止酯基同時被水解。化合物8發(fā)生催化氫化脫去芐基得到相應(yīng)的羧酸9。化合物9經(jīng)過胺化反應(yīng)得到所需的含有對位甲酰胺取代基的化合物10。該步驟也可使用偶聯(lián)試劑HATU/EDCI和NH4Cl進(jìn)行酰胺化,產(chǎn)率94%。再用鹽酸脫去Boc保護(hù)基得到片段B的鹽酸鹽化合物11,最后,將11進(jìn)行Fmoc保護(hù)得到所需的12。由于12在有機(jī)溶劑中的溶解性很差,后處理可以用乙酸乙酯等有機(jī)溶劑進(jìn)行多次洗滌,即可得到高純度的12。
接下來,嘗試測定此條路線合成的12的光學(xué)純度。發(fā)現(xiàn)由于12在常見的有機(jī)溶劑中溶解度很低,僅能少量溶解在DMSO中,難以使用HPLC測定其ee值。因此,在其羧酸端引入了一條聚乙二醇羧酸長鏈,得到了化合物14,大大提高了其溶解度,最終測得化合物14的ee值為97%,與化合物7的ee值接近,證明了此條路線的可靠性。
從對甲酸芐溴出發(fā),經(jīng)過不對稱烷基化反應(yīng),水解反應(yīng),胺化反應(yīng)等10步反應(yīng)合成了β2-AR別構(gòu)拮抗劑Cmpd15的關(guān)鍵中間體片段B。此條合成路線產(chǎn)率高,對映選擇性較好(97%ee)。該研究為Cmpd15及其類似物的合成提供了一條高效可靠的路線。
