白花香蓮解毒顆粒質(zhì)量標(biāo)準(zhǔn)研究
2020-12-03 04:04:48黃鵬邱華范春嬌蘇翠麗李家煥李旺蒙蔭杰何錦軼
中醫(yī)藥學(xué)報(bào) 2020年11期
黃鵬,邱華,范春嬌,蘇翠麗,李家煥,李旺,蒙蔭杰,何錦軼
(1.廣西中醫(yī)藥大學(xué),廣西 南寧 530001;2.廣西中醫(yī)藥大學(xué)第一附屬醫(yī)院,廣西 南寧 530023)
白花香蓮解毒顆粒是在壯醫(yī)“毒虛致病”理論指導(dǎo)下,由白花蛇舌草、黃花倒水蓮、三葉香茶菜、排錢草共4味壯藥組成,具有攻毒補(bǔ)虛、疏三道、通兩路、調(diào)三氣、平陰陽之效,是廣西中醫(yī)藥大學(xué)第一附屬醫(yī)院的臨床經(jīng)驗(yàn)方,至今已有十余年的臨床應(yīng)用歷史,主要用于治療慢性乙型肝炎,在提高HBV陰轉(zhuǎn)率、增加HBeAg血清學(xué)轉(zhuǎn)換率、改善生活質(zhì)量等方面具有顯著療效[1-9]。控制好藥品的質(zhì)量,以確保該制劑的臨床療效,這對(duì)于提高廣西慢性乙型肝炎治療水平及民族醫(yī)藥產(chǎn)業(yè)創(chuàng)新能力,推動(dòng)學(xué)科內(nèi)涵建設(shè)發(fā)展具有重要的示范性意義。本研究采用薄層色譜法對(duì)制劑中的白花蛇舌草和三葉香茶菜進(jìn)行定性鑒別,并檢查白花香蓮解毒顆粒的粒度、水分、溶化性、裝量差異和微生物限度,運(yùn)用高效液相色譜法對(duì)白花蛇舌草的特異性成分去乙酰車葉草酸甲酯的含量進(jìn)行測(cè)定。
1 儀器與試藥
1.1 儀器
ZF-20C型暗箱式自動(dòng)紫外分析儀(上海寶山顧村電光儀器廠),Agilent1260型高效液相色譜儀(美國Agilent公司),SQP型電子分析天平(賽多利斯科學(xué)儀器北京有限公司),HWS-28型電熱恒溫水浴鍋(上海齊欣科學(xué)儀器有限公司),101-2型電熱鼓風(fēng)干燥箱(北京科偉永興儀器有限公司),LRH-250-S型恒溫恒濕培養(yǎng)箱(韶關(guān)市泰宏醫(yī)療器械有限公司),JM-A6002型電子天平(余姚市紀(jì)銘稱重校驗(yàn)設(shè)備有限公司)。
1.2 試藥
白花香蓮解毒顆粒(廣西中醫(yī)藥大學(xué)附屬瑞康醫(yī)院藥物研發(fā)中心制備,批號(hào)分別為20171201、20171202、20171203),白花蛇舌草對(duì)照藥材(中國食品藥品檢定研究院,批號(hào):201605),齊墩果酸對(duì)照品(上海安譜實(shí)驗(yàn)科技股份有限公司,批號(hào):U4070025),去乙酰車葉草酸甲酯對(duì)照品(成都曼思特生物科技有限公司,批號(hào):MUST-17101706),乙腈為色譜純(美國Fisher公司,批號(hào):166579),其它試劑均為分析純。白花蛇舌草、排錢草、三葉香茶菜、黃花倒水蓮四味藥材均購于藥市,經(jīng)廣西中醫(yī)藥大學(xué)廖月葵教授鑒定。
2 方法與結(jié)果
2.1 薄層色譜鑒別
2.1.1 白花蛇舌草的薄層色譜鑒別
分別取白花香蓮解毒顆粒(批號(hào)分別為20171201、20171202、20171203)粉末適量,稱取約11 g,加入30 mL乙醇,超聲處理,濾紙過濾,蒸干濾液,殘?jiān)屑尤? mL無水乙醇,溶解,過濾,濾液蒸干,加入1 mL甲醇溶解殘?jiān)鳛楣┰嚻啡芤骸7Q取白花蛇舌草對(duì)照藥材3 g,加水50 mL,回流提取1 h,濾紙過濾,濾液濃縮至15 mL,加入30 mL乙醇,攪拌過濾,濾液濃縮至2 mL,加入適量的硅藻土,攪拌均勻,干燥,加入30 mL乙醇,過濾,蒸干濾液,再加入乙醇1 mL使殘?jiān)芙猓鳛閷?duì)照藥材溶液。另取去乙酰車葉草酸甲酯對(duì)照品,以濃度1 mg/mL配制溶液,作為對(duì)照品溶液。再取缺白花蛇舌草顆粒適量,與供試品同樣制備方法制成陰性對(duì)照溶液。根據(jù)薄層色譜法(通則0502)的試驗(yàn)[10],吸取上述四種溶液各5 μL,分別點(diǎn)狀于同一個(gè)硅膠G板上,以三氯甲烷-甲醇-水(5∶1.5∶0.1)作為展開劑,展開,取出,晾干,噴10%硫酸乙醇溶液,105 ℃加熱至斑點(diǎn)顯示清晰。在供試品色譜圖中,同一顏色的斑點(diǎn)顯示在對(duì)照品色譜圖的相應(yīng)位置。見圖1。
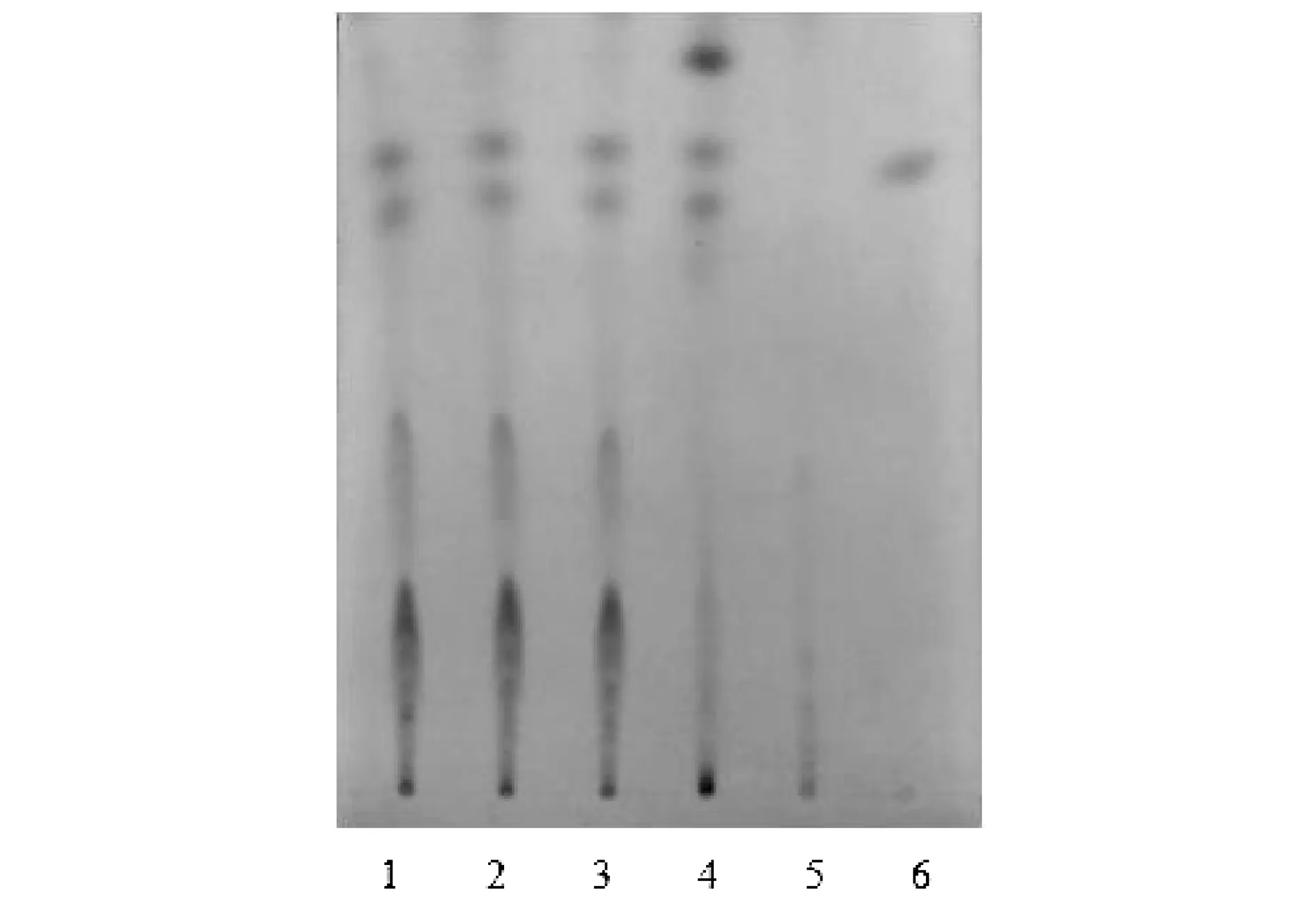
注:1~3為供試品溶液(顆粒批號(hào)分別為20171201、20171202、20171203);4為白花蛇舌草對(duì)照藥材溶液;5為白花蛇舌草陰性顆粒溶液;6為去乙酰車葉草酸甲酯對(duì)照品溶液。圖1 白花蛇舌草薄層色譜圖
2.1.2 三葉香茶菜的薄層色譜鑒別
分別稱取白花香蓮解毒顆粒(批號(hào)分別為20171201、20171202、20171203)3 g,加入10 mL甲醇,超聲處理,過濾,蒸干濾液,加入2 mL甲醇溶解殘?jiān)鳛楣┰嚻啡芤骸0瓷鲜鱿嗤苽浞椒ǎQ取三葉香茶菜對(duì)照藥材1 g,制備成對(duì)照藥材溶液。另取齊墩果酸對(duì)照品,以濃度0.5 mg/mL配制溶液,作為對(duì)照品溶液。再取缺三葉香茶菜與白花蛇舌草的顆粒3 g,與供試品同樣制備方法制成陰性對(duì)照溶液。根據(jù)薄層色譜法(通則0502)的試驗(yàn)[10],吸取上述四種溶液各7 μL,分別點(diǎn)狀于同一個(gè)硅膠G板上,以環(huán)己烷-丙酮-醋酸乙酯-甲酸(5∶2∶1∶0.1)為展開劑,展開,取出,晾干,10%硫酸乙醇溶液,105 ℃加熱至斑點(diǎn)顯示清晰。在供試品色譜圖中,同一顏色的斑點(diǎn)顯示在對(duì)照品色譜圖的相應(yīng)位置。見圖2。
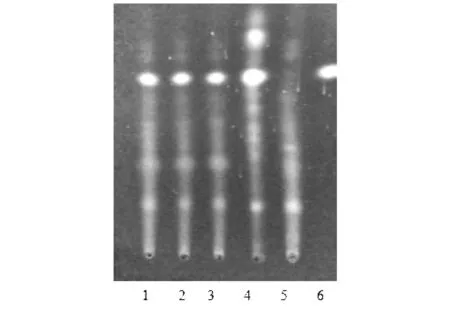
注:1~3為供試品溶液(批號(hào)分別為20171201、20171202、20171203);4為三葉香茶菜對(duì)照藥材溶液;5為三葉香茶菜陰性顆粒溶液;6為齊墩果酸對(duì)照品溶液。圖2 三葉香茶菜薄層色譜圖
2.2 白花香蓮解毒顆粒的粒度、水分、溶化性、裝量差異和微生物限度檢查
根據(jù)《中國藥典》[11](2015年版四部顆粒劑通則,0982第二法雙篩分法、0832烘干法、可溶顆粒溶化性測(cè)定法、裝量差異測(cè)定法、1105微生物計(jì)數(shù)法、1106控制菌檢查法及1107非無菌藥品微生物限度標(biāo)準(zhǔn)),分別測(cè)定三批中試顆粒的粒度、水分、溶化性、裝量差異和微生物限度,結(jié)果見表1,表2。

表1 白花香蓮解毒顆粒粒度、水分、溶化性、裝量差異檢查結(jié)果

表2 白花香蓮解毒顆粒微生物限度檢查結(jié)果
2.3 去乙酰車葉草酸甲酯的含量測(cè)定
2.3.1 色譜條件
色譜柱:Inertsil ODS-3(5 μm,4.6 mm×250 mm);流動(dòng)相:水-乙腈 (91∶9) ;檢測(cè)波長:238 nm;柱溫:30 ℃;流速:0.85 mL/min;進(jìn)樣量:5 μL。
2.3.2 標(biāo)準(zhǔn)品溶液的制備
取去乙酰車葉草酸甲酯標(biāo)準(zhǔn)品適量,精密稱定,加入9%乙腈水配制成以濃度1.014 mg/mL為標(biāo)準(zhǔn)溶液作為儲(chǔ)備液,精密吸取儲(chǔ)備液適量,加流動(dòng)相配制成濃度15.21 μg/mL的標(biāo)準(zhǔn)溶液,過0.45 μm微孔濾膜,得到標(biāo)準(zhǔn)品溶液。
2.3.3 供試品溶液的制備
取中試顆粒適量,研細(xì),取0.6 g,精密稱定,置于具塞錐形瓶中,精密加水20 mL,精密稱定,用超聲溶解,冷卻,用水補(bǔ)量,搖勻,過0.45 μm微孔濾膜,得到供試品溶液。
2.3.4 陰性對(duì)照溶液的制備
取缺白花蛇舌草的陰性顆粒適量,按2.3.3項(xiàng)下的方法配制陰性對(duì)照溶液。
2.3.5 系統(tǒng)適應(yīng)性試驗(yàn)
吸取標(biāo)準(zhǔn)品溶液,供試品溶液和陰性對(duì)照溶液分別按上述色譜條件進(jìn)樣5 μL,記錄色譜圖,結(jié)果如圖3~圖5所示。在標(biāo)準(zhǔn)品溶液去乙酰車葉草酸甲酯的出峰位置(a),供試品溶液在相同位置出峰且能較好分離(R>1.5),陰性對(duì)照溶液在同一位置沒有出現(xiàn)峰值。陰性對(duì)照樣品對(duì)去乙酰車葉草酸甲酯的含量測(cè)定無影響。
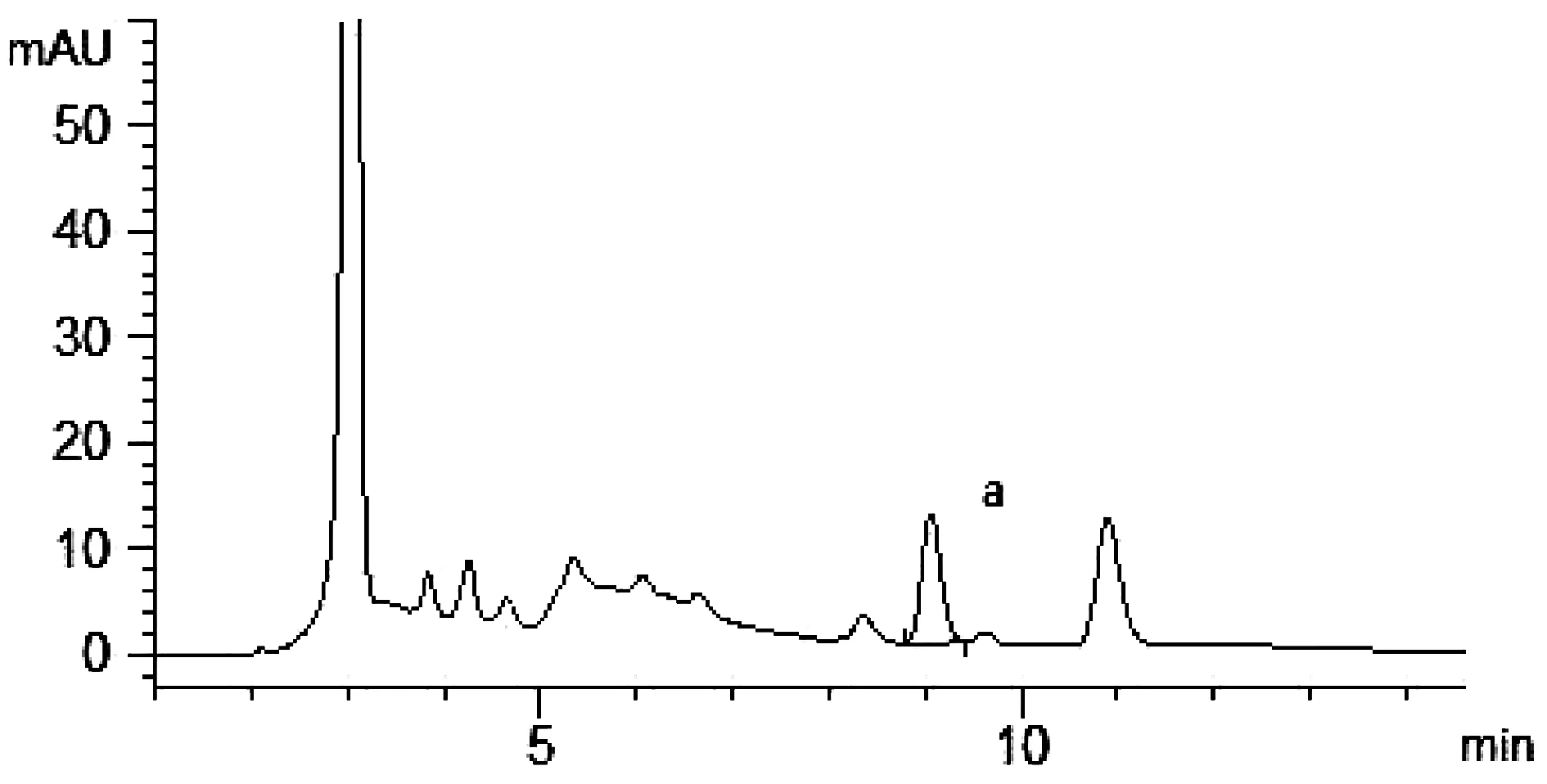
圖3 供試品溶液
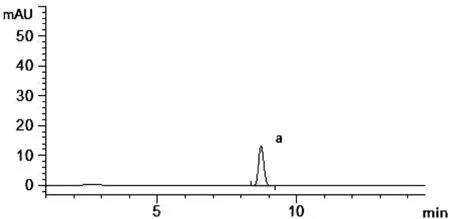
圖4 去乙酰車葉草酸甲酯對(duì)照品溶液
圖5 白花蛇舌草陰性對(duì)照溶液
2.3.6 線性關(guān)系考察
精密吸取標(biāo)準(zhǔn)儲(chǔ)備液去乙酰車葉草酸甲酯適量,加流動(dòng)相配制成濃度分別為5.070、10.14、15.21、20.28、30.42、40.56 μg/mL的標(biāo)準(zhǔn)品溶液,過0.45 μm微孔濾膜,按上述色譜條件測(cè)定。以標(biāo)準(zhǔn)品濃度(μg/mL)為橫向坐標(biāo)(X),峰面積積分值為縱向坐標(biāo)(Y)繪制標(biāo)準(zhǔn)曲線,回歸方程為Y=11.45X+1.016,r=0.999 9,去乙酰車葉草酸甲酯在5.07~40.56 μg/mL范圍內(nèi)線性關(guān)系良好。
2.3.7 精密度試驗(yàn)
精密吸取2.3.2項(xiàng)下的標(biāo)準(zhǔn)品溶液按上述色譜條件連續(xù)進(jìn)樣6次,并記錄所計(jì)算的峰面積的RSD為1.70%,表明本方法精密度良好。
2.3.8 穩(wěn)定性試驗(yàn)
稱取中試顆粒適量,按2.3.3項(xiàng)下的方法配制供試品溶液,在0~24 h內(nèi),每隔4 h進(jìn)樣5 μL,注入液相色譜儀,按上述色譜條件測(cè)定,記錄峰面積并計(jì)算RSD值。結(jié)果在24 h內(nèi),去乙酰車葉草酸甲酯峰面積的RSD值為0.50%,表明該溶液在24 h內(nèi)非常穩(wěn)定。
2.3.9 重復(fù)性試驗(yàn)
稱取中試顆粒適量,6份,按2.3.3項(xiàng)下的方法配制供試品溶液,分別吸取供試品溶液5 μL注入色譜儀,按上述色譜條件測(cè)定,記錄峰面積并計(jì)算RSD值。結(jié)果在含量測(cè)定條件下,去乙酰車葉草酸甲酯峰面積的RSD為0.96%,表明此方法重復(fù)性好。
2.3.10 加樣回收率試驗(yàn)
稱取已知含量的中試顆粒(含量為0.517 7 mg/g)約0.6 g,6份,精密稱定,加入適量的標(biāo)準(zhǔn)儲(chǔ)備液(1.014 mg/mL),按2.3.3項(xiàng)下的方法配制供試品溶液,分別吸取供試品溶液5 μL,按上述色譜條件測(cè)定,計(jì)算回收率和RSD值,結(jié)果見表3。
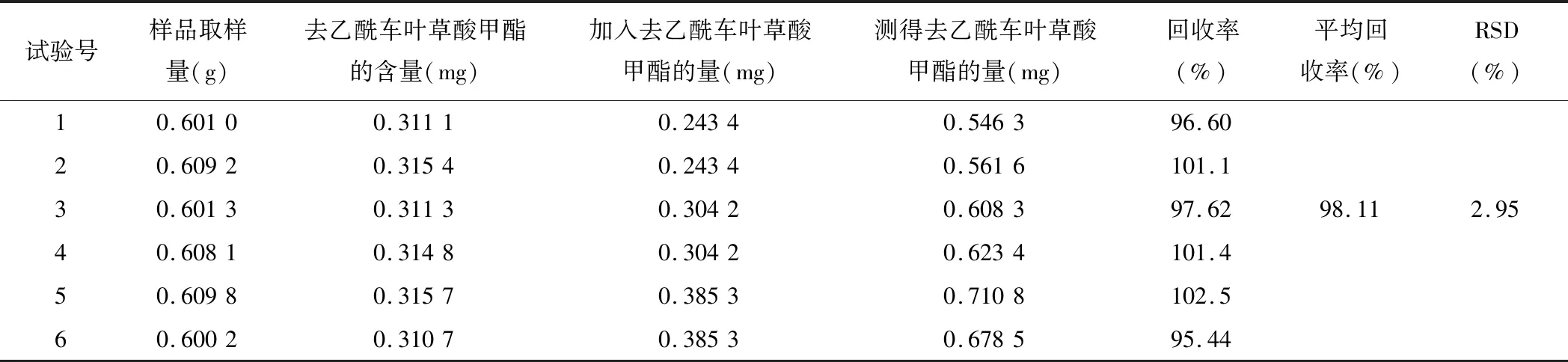
表3 加樣回收試驗(yàn)考察結(jié)果
2.3.11 樣品測(cè)定
精密吸取對(duì)照品溶液和供試品溶液各5 μL,按上述色譜條件對(duì)三批中試顆粒進(jìn)行含量測(cè)定,每批樣品平行測(cè)定3份,結(jié)果三批中試顆粒的含量測(cè)定結(jié)果變化幅度不大。含量限度以批次最低含量(0.489 7 mg/g)下調(diào)20%計(jì),確定顆粒中有效成分的最低含量限度,即每袋(14 g)去乙酰車葉草酸不低于5.5 mg。見表4。
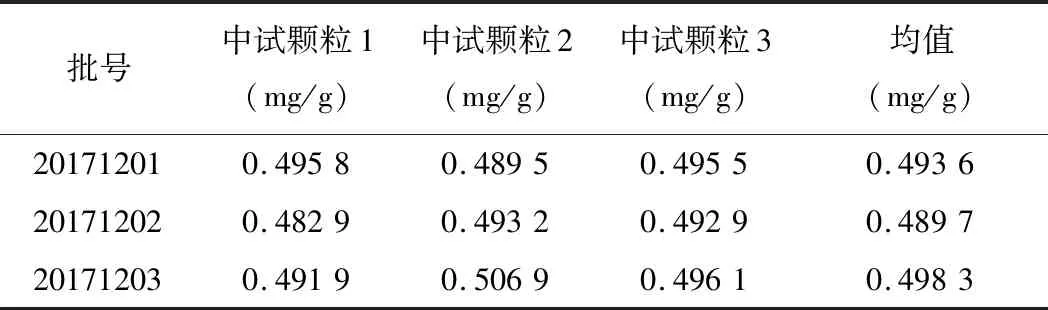
表4 中試顆粒去乙酰車葉草酸甲酯含量測(cè)定結(jié)果
3 討論
3.1 薄層鑒別
本試驗(yàn)所確定的薄層色譜法可鑒別出白花香蓮解毒顆粒中白花蛇舌草和三葉香茶菜,方法簡單、穩(wěn)定,可作為白花香蓮解毒顆粒的定性鑒別方法。白花蛇舌草主要含有蒽醌類、黃酮類、萜類等化學(xué)成分[11],參照《中國藥典》2015年版四部花紅顆粒以及清腎顆粒中的方法對(duì)白花蛇舌草進(jìn)行定性鑒別,結(jié)果表明,薄層色譜清晰,重現(xiàn)性好,專屬性強(qiáng)。三葉香茶菜來源于唇形科香茶菜屬植物的干燥全草,主要含有齊墩果酸、熊果酸等有效成分[12],其中齊墩果酸、熊果酸常為三葉香茶菜質(zhì)量控制的指標(biāo),兩者互為同分異構(gòu)體,薄層鑒別很難分離這兩個(gè)成分。本課題組選擇齊墩果酸作為三葉香茶菜薄層鑒別的對(duì)照品,因白花蛇舌草中也含有齊墩果酸,故以雙陰顆粒制備陰性對(duì)照溶液,以免對(duì)薄層結(jié)果進(jìn)行干擾。廣西壯族自治區(qū)瑤藥材質(zhì)量標(biāo)準(zhǔn)(第一卷)中對(duì)排錢草進(jìn)行定性鑒別時(shí),對(duì)照品為β-谷甾醇,而白花香蓮解毒顆粒中白花蛇舌草和三葉香茶菜均含有β-谷甾醇,且其為脂溶性成分,故此方法不適用于排錢草的定性鑒別。本課題組曾考慮用排錢草的成分N,N-二甲基色胺、5-甲氧基-N,N-二甲基色胺進(jìn)行薄層鑒別,但由于實(shí)驗(yàn)條件的限制,較難自制出以上兩種成分,故放棄此方法進(jìn)行薄層鑒別。本課題組參照三草護(hù)肝膠囊薄層鑒別中的方法,對(duì)排錢草和黃花倒水蓮進(jìn)行薄層鑒別時(shí),發(fā)現(xiàn)陰性顆粒在相應(yīng)的位置干擾較大,曾多次試驗(yàn)都無法去除陰性顆粒斑點(diǎn)的干擾,且兩者薄層鑒別的文獻(xiàn)記載有限,故本試驗(yàn)未能建立排錢草和黃花倒水蓮薄層鑒別的方法。
3.2 含量測(cè)定
本試驗(yàn)采用主藥的特異性成分去乙酰車葉草酸甲酯作為白花香蓮解毒顆粒質(zhì)量控制的指標(biāo),通過查閱《中國藥典》2015年版四部花紅顆粒中關(guān)于去乙酰車葉草酸甲酯含量測(cè)定的方法,建立了測(cè)定去乙酰車葉草酸甲酯含量的方法,從而快速、簡便、有效的控制了該制劑的質(zhì)量。
