川崎病急性期患兒血清對人臍靜脈血管內皮細胞氧化應激、增殖及凋亡的影響
2020-12-02 03:40:12胡景偉牛紅艷薩初然貴高崇華楊淑蘭
山東醫藥 2020年32期
胡景偉,牛紅艷,薩初然貴,高崇華,楊淑蘭
內蒙古醫科大學赤峰臨床醫學院,內蒙古赤峰024000
川崎病(KD)又稱為皮膚黏膜淋巴結綜合征,是一種以全身中、小動脈炎性病變為主要病理變化的急性熱性發疹性疾病,冠狀動脈病變(CAL)是KD最嚴重的并發癥[1]。研究發現,KD患者存在不同程度的血管內皮細胞損傷[2],表現為細胞剝脫、裸露、間裂隙增大、穿孔等,這些損傷與CAL并發癥密切相關,即使在靜脈應用丙種球蛋白(IVIG)治療后仍可持續存在[3]。近年來,氧化應激(OS)在血管性疾病病理過程中發揮的作用日益受到重視。目前常用的OS相關指標主要包括丙二醛(MDA)和超氧化物歧化酶(SOD),前者反映細胞受自由基攻擊的損傷程度,后者是一種強抗氧化劑。有研究顯示,外周血淋巴細胞凋亡異常與KD發病有關,但關于血管內皮細胞凋亡與KD關系的研究尚不多見。2017年1月~2019年10月,本研究觀察了KD急性期患兒血清對人臍靜脈血管內皮細胞(HUVEC)氧化應激、增殖和凋亡影響,現報告如下。
1 材料與方法
1.1 血清、細胞與主要材料 選擇2017年1月~2018年1月于我院住院治療的KD急性期患兒12例,男7例、女5例,年齡1歲5個月~6歲11個月,均符合2004年美國兒科學會和心臟病學會制訂的診斷標準,病程均在1周以內,未經過IVIG治療。選取同期在本院兒科門診體檢的健康兒童10例作對照,男6例、女4例,年齡2歲1個月~6歲10個月。檢查證實肝腎心功能正常,無基礎心血管疾病,無感染性及免疫性疾病。在經醫院醫學倫理委員會批準、研究對象監護人知情同意的情況下,抽取KD患兒及健康兒童外周靜脈血2 mL,EDTA抗凝,1 500 r/min離心5 min,收集上清液,-80 ℃冰箱保存。HUVEC(上海中喬新舟生物科技有限公司),RPMI-1640培養基(美國Hyclone公司),CCK-8溶液(中國BioSharp公司),MDA、SOD ELISA試劑盒(武漢百仟度生物科技有限公司),FACSCalibur流式細胞儀(美國BD公司),DCFH-DA粉劑(美國Sigma公司),RNA提取液(美國Invitrogen公司),RevertAid First Strand cDNA Synthesis Kit(美國Thermo公司),FastStart Universal SYBR Green Master(美國Roche公司),熒光定量PCR儀(美國ABI公司),引物由武漢百仟度生物科技有限公司合成(引物序列見表1),兔抗人Bcl-2一抗(美國CST公司),兔抗人Caspase-3一抗(英國Abcam公司),兔抗人Bax一抗(英國Abcam公司),山羊抗兔HRP標記二抗(美國KPL公司)。

表1 Bax、Caspase-3、Bcl-2基因引物序列
1.2 HUVEC培養和分組 HUVEC培養于RPMI-1640培養基中,將培養皿置于含5%CO2的37 ℃恒溫培養箱。根據處理條件的不同將HUVEC細胞分為正常組和KD組,分別加入15%健康兒童血清和15% KD患者血清培養24 h。
1.3 OS相關指標檢測
1.3.1 MDA、SOD含量檢測 收集兩組HUVEC懸液置于試管中,采用ELISA法檢測MDA和SOD。
1.3.2 活性氧(ROS)陽性細胞百分比測算 將處于對數生長期的HUVEC接種于6孔板內,置于5%CO2、37 ℃條件培養箱中培養24 h。按“1.2”方法分組處理細胞,培養24 h后加入終濃度為10 μmol/L的DCFH-DA,37 ℃下避光孵育30 min,每隔3~5 min混勻,使探針和細胞充分作用,離心去上清,洗滌并重懸細胞后上流式細胞儀測算ROS陽性細胞百分比(激發光488 nm,發射光525 nm)。每組設3個樣本。重復3次。
1.4 細胞增殖能力觀察 采用CCK-8法。將對數生長期的HUVEC用胰蛋白酶消化,按1×104/孔接種于96孔板,每孔加100 μL,置于5%CO2、37 ℃條件培養箱培養24 h。按“1.2”方法分組處理細胞,培養24 h后每孔補加10 μL的CCK-8試劑,孵育2 h,用酶標儀測定在450 nm處的光密度(OD)值,表示細胞增殖能力。每組樣本設3個空孔,重復3次。
1.5 細胞凋亡相關基因及蛋白檢測
1.5.1 凋亡相關基因檢測 按試劑盒說明提取兩組HUVEC的總RNA,按逆轉錄合成試劑盒說明書合成cDNA,將合成的cDNA與引物序列等配制成反應體系,采用RT-PCR法檢測Bax、Caspase-3、Bcl-2 mRNA,以GAPDH作為內參。以2-ΔΔCt表示目的基因相對表達量。
1.5.2 凋亡相關蛋白檢測 采用Western blotting法。以β-actin為內參,Image J軟件分析蛋白條帶灰度值作為目的蛋白相對表達量。
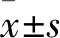
2 結果
2.1 兩組OS相關指標比較 KD組MDA水平、ROS陽性細胞百分比高于正常組,MDA水平低于正常組(P均<0.05)。見表2。
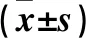
表2 兩組OS相關指標比較
2.2 兩組細胞增殖能力比較 KD組、正常組細胞增殖能力分別為0.54±0.01、0.84±0.03,KD組細胞增殖能力低于正常組,兩組差異有統計學意義(P<0.01)。
2.3 兩組凋亡相關基因及蛋白表達比較 KD組Bax、Caspase-3 mRNA和蛋白相對表達量高于正常組,Bcl-2 mRNA和蛋白相對表達量低于正常組(P均<0.05)。詳見表3。
表3 兩組凋亡相關基因及蛋白表達比較
3 討論
KD是一種主要累及中小動脈的全身血管炎性疾病,本病是否發生CAL是決定患者長期預后的關鍵因素。研究發現,CAL與血管內皮細胞功能障礙有關[4],因此,研究KD患者血管內皮細胞損傷的發生機制具有重要意義。造成KD血管內皮損傷的機制有很多,本研究從OS、細胞增殖和凋亡的角度,通過體外實驗研究探討KD患兒血管內皮細胞損傷的機制。
OS是指機體在遭受有害刺激時,體內ROS產生過多,氧化程度超過氧化物的清除速度,導致組織損傷。研究顯示,OS通過細胞信號轉導、蛋白質變性、脂質過氧化反應等途徑引起細胞損傷及壞死,促進淋巴細胞和單核巨噬細胞遷移,從而導致血管內皮細胞的通透性增加、引起細胞的壞死和凋亡[5]。Kaneko等[6]發現,KD急性期患者ROS水平明顯高于發熱患者,而KD患者在應用IVIG后與應用IVIG之前比較,ROS明顯降低,提示OS參與KD的發生。還有研究發現,KD急性期患者血清8-異構前列腺素、MDA和H2O2明顯升高[7,8],尤其是在伴有CAL的患者,這種改變更加明顯[9]。8-異構前列腺素是DNA氧化應激損傷的產物之一,MDA是膜脂過氧化產物,兩項指標改變均提示OS參與了KD患者CAL的發生。有學者發現,KD患者血清OS相關指標明顯高于健康兒童,經過IVIG治療后OS指標明顯下調,但是仍然高于健康兒童[10],提示OS持續存在,這可能是部分KD患者即使應用IVIG治療仍然發生CAL的原因[11]。Xu等[12]發現,小檗堿可通過抑制OS保護KD患者冠狀動脈內皮細胞,減輕損傷。本研究結果顯示,KD急性期患兒血清作用于HUVEC后導致參與OS的ROS和OS產物MDA水平明顯升高,抗氧化應激反應的SOD水平明顯降低,與前述結果一致。這提示KD患者存在氧化/抗氧化系統失衡,而且血管內皮細胞是OS的作用靶細胞,相關具體機制尚需進一步研究,可能與抑制Nrf2和PI3K/AKT信號通路有關[13]。
研究發現,KD患者外周血單個核細胞誘導分化的內皮組細胞與正常兒童組比較,其增殖能力、遷移能力和貼壁能力均顯著下降[14]。有學者將HUVEC經正常血清、發熱患兒血清、無CAL和合并CAL的KD患者血清處理后,應用MTT法檢測細胞增殖情況,結果顯示,經KD血清干預后的細胞活性低于正常組和發熱組,且合并CAL組低于無CAL組,提示KD血清能夠抑制HUVEC增殖[15]。本研究采用CCK-8法檢測細胞增殖能力,結果發現KD組細胞增殖能力明顯低于正常組。這提示KD急性期血清對HUVEC增殖具有抑制作用,與前述研究結果相一致,具體機制可能與TNF-α有關。有學者發現TNF-α可以通過PI3K/AKT/eNOS信號途徑損傷內皮祖細胞,而在KD患者血清內TNF-α明顯升高[16]。還有研究認為,KD患兒血清對HUVEC增殖的抑制作用可能與miR-27b通過影響轉化生長因子-β的活性實現的[17]。
劉麗莎等[15]發現,經KD患兒血清作用后,HUVEC的凋亡率增加,與對照組相比差異有統計學意義,且CALs組凋亡率高于非CALs組。本研究檢測了兩組細胞中凋亡相關基因和蛋白(Bcl-2、Bax、Caspase-3)表達,發現KD組促進凋亡的Bax、Caspase-3 mRNA和蛋白相對表達量高于正常組,而抑制凋亡的Bcl-2 mRNA和蛋白表達低于正常組,提示KD患兒血清可導致HUVEC凋亡增加,其具體機制需進一步研究。研究顯示,KD患者血中Caspase-4基因表達與健康對照組相比顯著升高,NF-κB信號通路相關因子表達也發生變化,且兩者存在線性關系,提示嬰幼兒在受到環境因素和遺傳因素影響后,NF-κB信號通路激活后被釋放進入細胞核內,引起Caspase-4基因表達變化,進而導致KD的發生[18]。Wu等[19]的研究表明,KD血清可以導致HUVEC凋亡,這一過程與miR-186升高導致MAPK信號通路激活有關。還有研究發現,經KD血清處理過的單核巨噬細胞可以激活HMGB1/RAGE/組織蛋白酶B信號途徑,導致NLRP3炎癥小體活化,進而引起HUVEC壞死[20]。
綜上所述,HUVEC經KD急性期患兒血清處理后,細胞氧化應激損傷增強,細胞增殖抑制而凋亡增加,這些異常變化可能與KD的發生密切相關。但是,KD患者血清中何種成分導致上述變化及其相應機制尚需進一步研究。
猜你喜歡
體育科技文獻通報(2022年3期)2022-05-23 13:46:54
中老年保健(2021年3期)2021-08-22 06:50:04
天津外國語大學學報(2021年3期)2021-08-13 08:32:18
遼金歷史與考古(2021年0期)2021-07-29 01:06:54
昆明醫科大學學報(2021年1期)2021-02-07 01:06:36
現代臨床醫學(2021年1期)2021-01-26 00:56:02
中華養生保健(2020年4期)2020-11-16 01:31:40
中西醫結合肝病雜志(2020年2期)2020-10-27 02:18:50
科技傳播(2019年22期)2020-01-14 03:06:54
民用飛機設計與研究(2019年4期)2019-05-21 07:21:24
