閥切換-離子色譜法測定天然氣水合物試采產出水中的氯離子和硫酸根
2020-11-09 06:40:30劉紀勇陸紅鋒薩日娜
分析儀器 2020年5期
關鍵詞:分析
劉紀勇 陸紅鋒 薩日娜 劉 玲
(廣州海洋地質調查局,廣州 510760)
天然氣水合物被科學家認為是一種儲量豐富的清潔能源,其資源量是全球石油天然氣和煤炭等化石能源資源量總和的兩倍[1]。最近幾年對于海域天然氣水合物的試采研究工作空前活躍,隨著天然氣水合物基礎研究的不斷深入,目前已進入試開采階段,全球已有4個國家開展了天然氣水合物試采,包括加拿大、美國、日本及中國。其中對海域天然氣水合物進行試采的國家僅日本和中國[2]。
2017年,我國對南海神狐海域進行了首次試采,取得了持續產氣時間最長,產氣總量最大等突破性成果[3,4]。在試采過程中,對產出水中氯離子、硫酸根進行了離子色譜法實時監測,由于水合分解對周圍孔隙水具有淡化效應,通過氯離子和硫酸根濃度隨試采時間的變化,可以間接估算水合物的分解情況。但試采過程中為了抑制二次水合物生成,會向生產井中加入大量乙二醇溶液,這就使得現場測試過程中所取的水樣中含有一定比例的乙二醇。乙二醇與水能以任意比例互溶,分離難度大,給離子色譜法測定產出水中氯離子、硫酸根帶了極大的困難。
目前氯離子、硫酸根的檢測方法常用的方法主要有:濁度法[5-8]、分光光度法[9-12]、滴定法[13-16]、流動注射法[17]、離子色譜法[18-24]等。簡單化學濁度法、分光光度法和滴定法實驗過程繁瑣、時間長,一些化學試劑對身體有害;流動注射技術不能進行多元素同時測定分析;而離子色譜法自從1975 年被Small 等[25]提出之后就成為一種有效的分離測定方法,近年來隨著離子色譜技術的快速發展, 其在多種陰陽離子同時分離測定方面應用更為廣泛。多數是測定簡單基質中陰陽離子,閥切換-離子色譜對于分析高濃度復雜基質中氯離子、硫酸根,如海水、孔隙水,特別是含有機組分的海水報道較少。閥切換-離子色譜分析技術在醫藥、食品領域有廣泛報道,但基本都是利用閥切換對待測離子進行富集,用如張婷婷等[26]開展了閥切換-離子色譜法測定分析純硫酸鈉中痕量氯離子,由于經歷了大量時間的樣品富集,分析時間長達38分鐘/樣。因此,本實驗設計了閥切換-離子色譜系統,建立了測定含乙二醇試采產出水中氯離子、硫酸根的方法并對方法的精密度、準確度、加標回收率等參數進行分析。
1 實驗部分
1.1 儀器與試劑
儀器:美國Thermo Fisher Scientific公司ICS 5000+型通用離子色譜儀, Dionex IonPac AS18-Fast陰離子色譜柱,Dionex IonPac AG18保護柱,AERS 500抑制器, Dionex IonPac UTAC-ULP1捕獲柱,電導檢測器,5μL定量環,AXPC泵,AS-AP自動進樣器及其配套軟件。
試劑:所用試劑均為分析純,所有溶液均用Mill-Q超純水配制,氯離子、硫酸根混合標準溶液濃度為1000 mg/L。
1.2 樣品預處理
用聚丙烯塑料瓶從試采產水管道取得水樣后,用5mL一次性注射器取約3mL水樣進行二級過濾,一級過濾和二級過濾的濾膜孔徑分別為0.45μm和0.2μm,樣品過濾至潔凈玻璃管中待用。
1.3 離子色譜分析條件
淋洗液:25.5mmol/L NaOH,經0.20μm濾膜過濾并脫氣,流速1.0mL/min,AXPC洗脫液流速1.0mL/min,色譜柱溫度23℃,閥切換時間為進樣后1min。
2 結果與討論
2.1 離子色譜分析系統建立
天然氣水合物試采過程中,為了抑制二次水合物形成,會向生產井中加入大量乙二醇溶液,使得現場測試過程中所取的水樣中含有一定比例的乙二醇溶液,由于乙二醇與水能以任意比例互溶,且分離難巨大,為現場離子色譜法測定試產出水中氯離子、硫酸根帶來了極大的困難。主要體現為乙二醇屬有機溶劑,在進入離子色譜系統后會對離子色譜保護柱、分析柱及抑制器產出嚴重影響,隨著測試進行,離子色譜會出現色譜柱柱效下降、基線漂移、保留時間變化等,導致測試的精密度、準確度下降,也會對離子色譜硬件產出不可逆的損害。因此針對此難點設計了一種閥切換系統,并制定一個在線樣品前處理方案,如圖1所示。
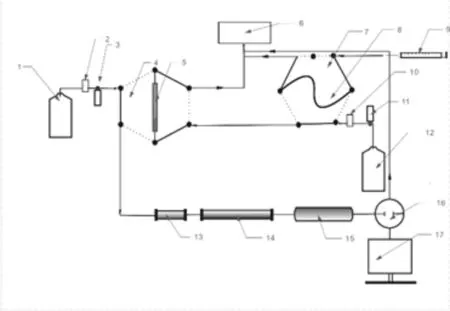
圖1 離子色譜分析系統示意圖1.淋洗液;2.1號脫氣裝置;3.活塞泵;4.1號六通閥;5.陰離子捕獲柱;6.廢液池;7.2號六通閥;8.定量環;9.進樣針;10.2號脫氣裝置;11.AXPC泵;12.超純水;13.保護柱;14.分離柱;15.抑制器;16.電導檢測器;17.記錄終端
該方案是在樣器進入離子色譜系統前,將目標離子保留在一根色譜捕獲柱上,而乙二醇不被保留,被超純水沖至廢液中。再用淋洗液將被保留的目標離子帶進色譜系統進行分析,實現了在線去除乙二醇的目的,從而使離子色譜能夠長期穩定的運行。
2.2 定量環的選擇
對5μL、10μL、25μL3種規格定量環在上述離子色譜條件下進行了多次實驗,結果表明,3種定量環均能獲得良好的線性關系,但考慮到天然氣水合物試采過程中需要測量大量的樣品,小的進樣量可以減小樣品的稀釋倍數,從而減小稀釋液的消耗。因此選擇5μL作為樣品的進樣量。
2.3 淋洗液濃度的選擇
淋洗液濃度的大小直接影響出峰時間,合理的淋洗液溶液可以極大的提高測試效率,分別選擇22.0mmol/L、25.2 mmol/L、31.0 mmol/L的氫氧化鈉溶液作為淋洗液進行了多組實驗發現,采用低濃度淋洗液時,分析時間過長,不利于試采現場分析,采用高濃度的淋洗液時硫酸根會被溴離子干擾,因此最終選擇25.2 mmol/L作為淋洗液濃度,可以在較短的時間內獲得良好的峰形。
2.4 閥切換時間的先擇
自動進樣后,用超純水將樣品帶至捕獲柱,待測離子被保留在捕獲柱中,乙二醇濃度不被保留而直接被超純水洗脫至廢液中,待洗脫完全后,閥切換至淋洗系統,淋洗液將待測離子洗脫至分析系統中進行檢測。研究發現,超純水洗脫1min后可將捕獲柱中的乙二醇全部沖洗至廢液。所以閥切換時間設為1min。
2.5 標準工作曲線、精密度及檢測限
將標準溶液稀釋成10.00、20.00、50.00、80.00、100.00mg/L的混合標準溶液,依次進樣分析后所得圖譜如圖2所示。以峰面積為縱坐標,標準溶液濃度為橫坐標,建立了標準工作曲線,同時對50、100 mg/L的標準溶液進了5次平行測定,計算了方法的精密度,色譜圖如圖3所示。標準曲線回方程、檢測限和相對標準偏差,如表1所示。
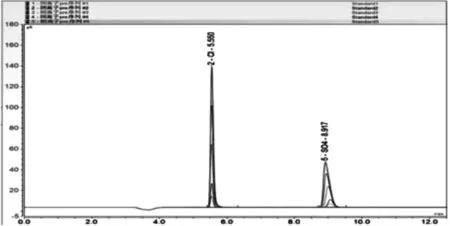
圖2 Cl-、SO42-混合標準樣品系列色譜圖
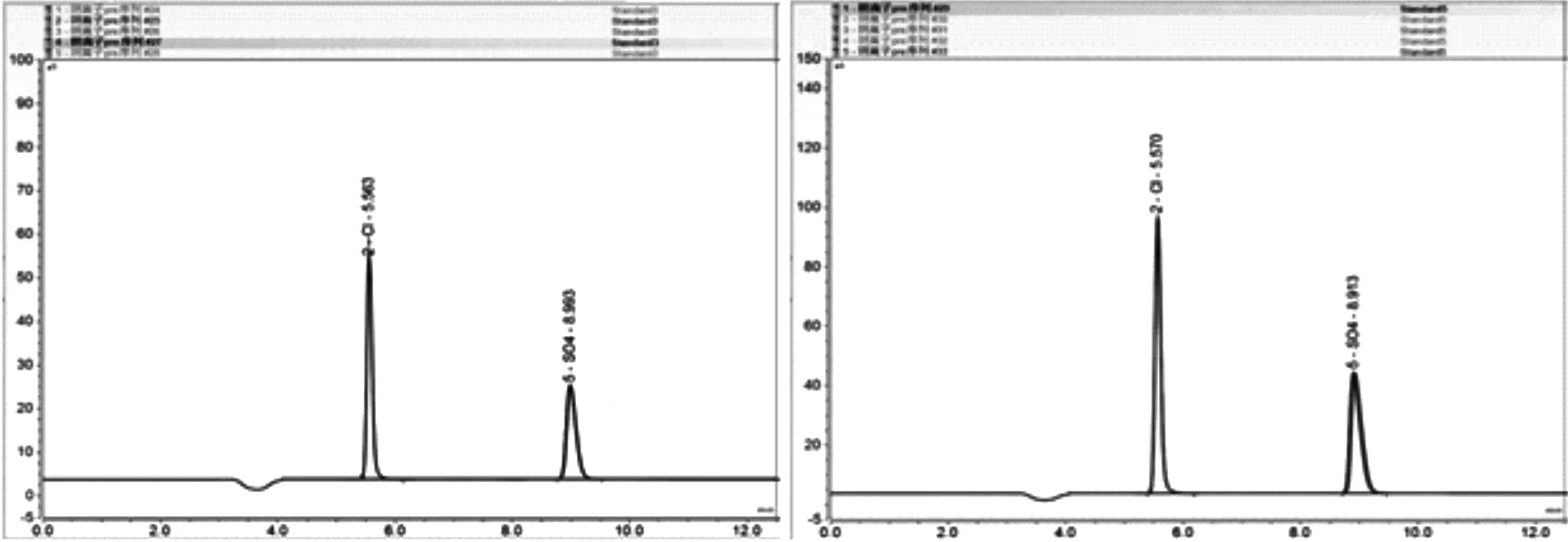
圖3 50、100 mg/L Cl-、SO42-混合標準樣品5次平行測定色譜圖

表1 校正曲線、相關系數、檢測限和相對標準偏差
從圖2中可看出,Cl-、SO42-保留時間分別為 5.550 min、8.917 min,具有良好的分離度和較短的分析時間,葛璐等[27]采用碳酸鹽淋洗液體系,分析時間為15min。從圖3中可以看出方法具有良好的重復性,在對50、100mg/L標準溶液5次平等測試過程中,峰形基本完全重合,從表1中也可看出,方法的線性較好,相關系均大于0.999,檢出限良好,分別為0.0085mg/L和0.0214mg/L,相對標準偏差均小于2%,精密度較好。
2.6 樣品測試
將玻璃管中過濾后的樣品取0.5mL至100mL容量瓶中稀釋至刻度,將稀釋后的樣品裝入進樣瓶中放入AS-AP自動進樣器進行測試。對樣品進行了5次平等進樣,測試結果如圖4所示。從圖中可看出經過200倍稀釋后,圖譜的峰高落在標曲線范圍內,平行測定圖譜重合性較好,Cl-、SO42-測試的標準偏差(RSD)分別為0.62%、0.052%,具有良好精密度,如表2所示。
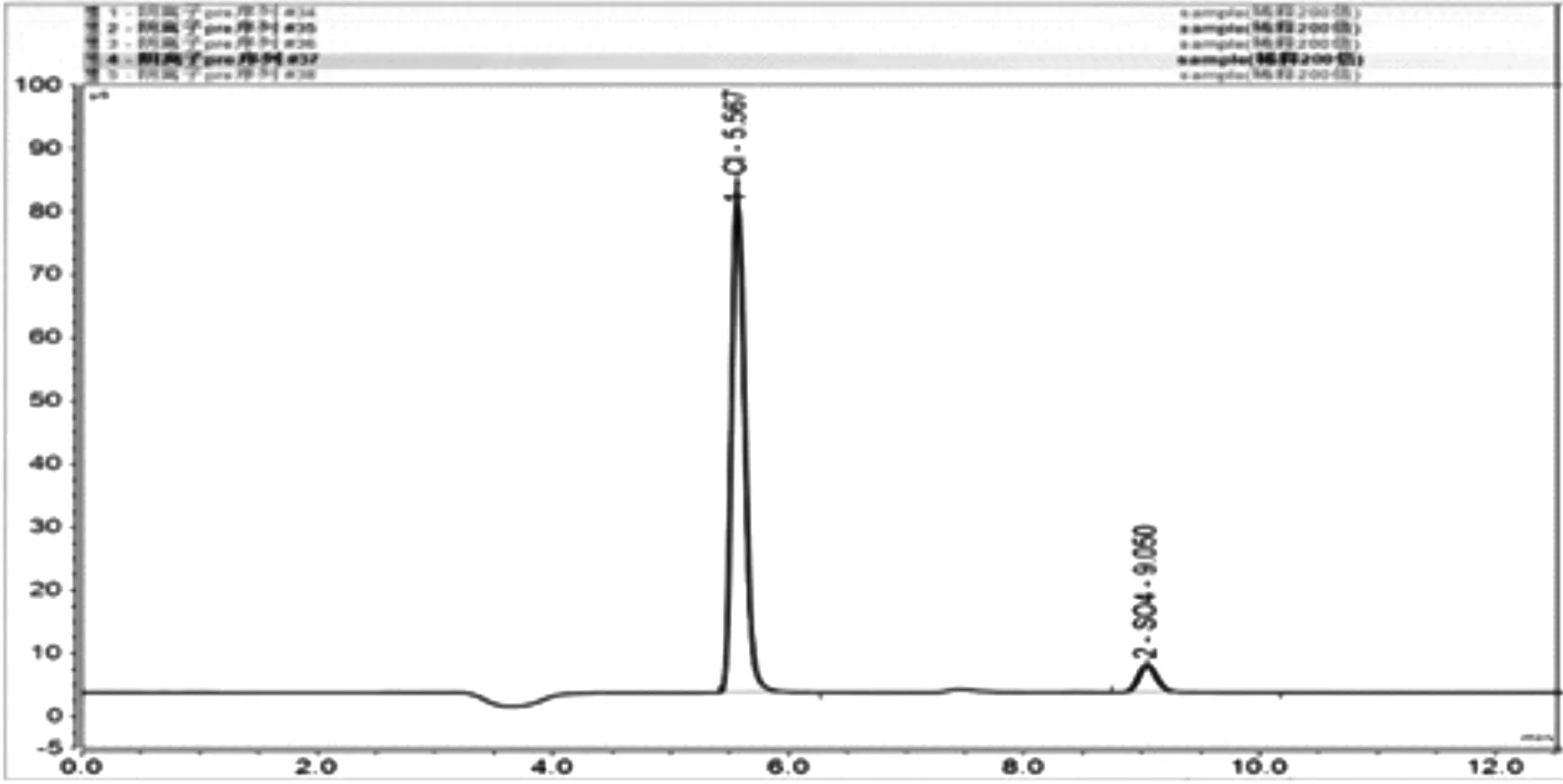
圖4 稀釋200倍的樣品5次平等測定色譜圖

表2 樣品平行測定結果
2.7 加標回收率測定
將稀釋后的樣品與80mg/L的Cl-、SO42-混合標準溶液進行等體積混合,混合均勻后進了5次平行測定,測試結果如圖5及表3所示。
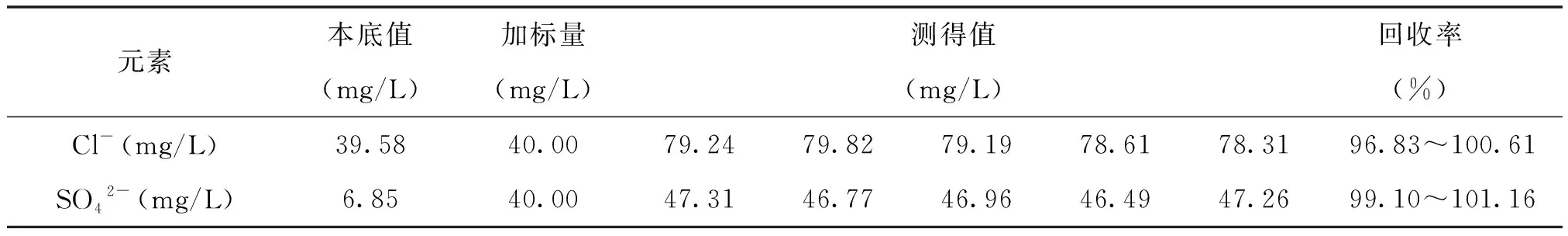
表3 回收率試驗結果
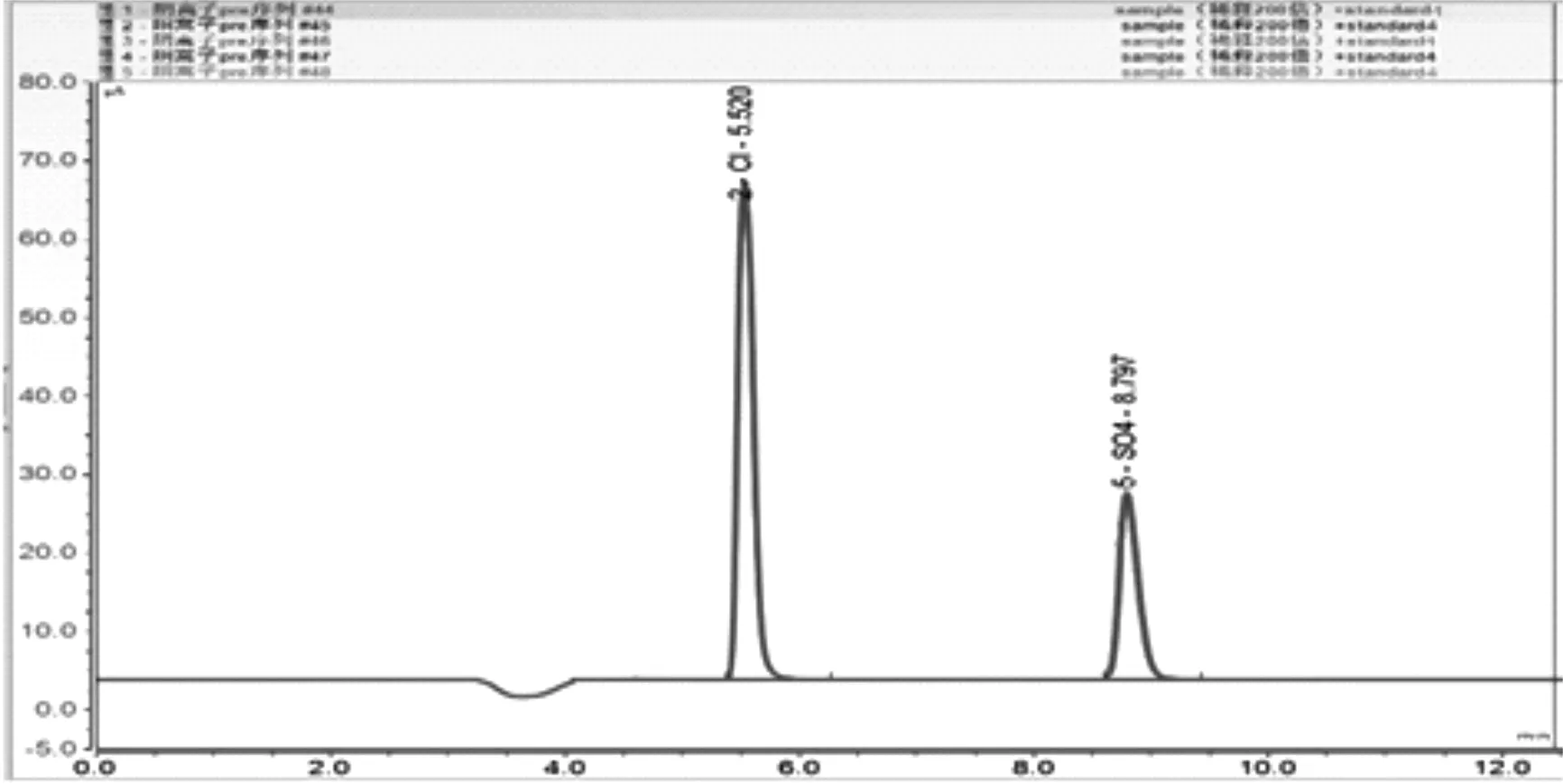
圖5 回收率5次平等測定色譜圖
從表3中可看出,氯離子和硫酸根的加標回收率分別為96.83~10.61%和99.10~101.16%,表明該方法的準確度較好,符合樣品分析要求。
3 結論
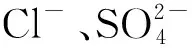
4 應用
猜你喜歡
現代畜牧科技(2021年9期)2021-10-13 06:39:14
民用飛機設計與研究(2020年4期)2021-01-21 09:15:02
電子制作(2018年18期)2018-11-14 01:48:24
山東工業技術(2016年15期)2016-12-01 05:31:22
當代經濟研究(2016年5期)2016-12-01 03:12:05
現代農業(2016年5期)2016-02-28 18:42:46
出版與印刷(2016年3期)2016-02-02 01:20:11
中國中醫藥現代遠程教育(2014年11期)2014-08-08 13:23:44
華北水利水電大學學報(社會科學版)(2014年3期)2014-04-16 04:38:31
終身教育研究(2014年5期)2014-02-28 01:23:06
