分子結拉伸與界面識別: 破解4, 4′-二吡啶分子結拉伸過程中高低電導之謎*
2020-11-06 03:23:24索雨晴劉然孫峰牛樂樂王雙雙劉琳李宗良
物理學報 2020年20期
關鍵詞:體系
索雨晴 劉然 孫峰 牛樂樂 王雙雙 劉琳 李宗良
(山東師范大學物理與電子科學學院, 濟南 250358)
1 引 言
20世紀80年代, 掃描隧道顯微鏡(STM)技術[1,2]的產生使得人類第一次能夠在單原子層次上觀察物質的表面形貌, 這為進一步控制單個原子或分子并構建單分子功能器件提供了條件. 隨后相繼出現的原子力顯微鏡(AFM)[3,4]、力學可控裂結(MCBJ)技術[5,6]以及各種單分子技術的組合使用[7,8], 促使納電子學與分子電子學在近二十多年得到快速發展[3,9?13]. 特別是自1997年Reed等[5]首次從實驗上實現單分子結的組裝與測量以來, 國內外科研團隊在分子開關[12?16]、分子整流器[17?21]、分子存儲器[22,23]、分子場效應管[24?28]、分子傳感器[29?31]等方面開展了大量的設計與研究工作, 并且發現了許多具有優異特性的功能分子器件. 然而, 到目前為止仍沒有設計出成熟的能夠進入應用領域的分子器件, 原因在于目前構建具有穩定性能的分子器件仍然存在許多亟待突破的技術挑戰[32?34].
雖然分子電子學的研究目標是在分子層次上構建功能電子器件并組裝邏輯電路, 但是伴隨分子電子學發展起來的單分子結制備技術為人們在單原子/分子層次上理解體系的物理過程、物理性質及其相互作用機理提供了強有力的技術手段[35].在分子電子學實驗中需要組裝單分子結并對其電輸運性質進行測量, 因此獲取到的是單分子層次的電輸運信號, 這是分子電子學實驗的獨特優勢. 盡管測量到的單分子電流信號不一定是滿足功能器件特定要求的電輸運信號, 但信號中包含了豐富的單分子、電極界面結構及其相互作用的豐富信息[8,36?38]. 到目前為止, 在分子電子學的研究中已經對各種不同類型的分子進行了測量, 從分子骨架上看有共軛分子[37?40]、非共軛分子[41?43]等, 從末端連接基團上分類有硫醇/酚末端分子[43?47]、氨基末端分子[6,45?48]、吡啶末端分子[47,49?54]等. 不同的分子, 特別是不同末端連接基團的分子, 與電極之間存在不同的相互作用, 并且表現出不同的電輸運特征[47,50,53,55]. 因此, 從理論上探索分子結體系的電輸運特性跟體系結構特別是體系結構變化過程之間的對應關系, 可以為實驗探測和理解分子體系的結構及其物理過程提供理論與數據支持.
從近二十幾年的實驗測量文獻報道中發現, 以吡啶基為末端的分子是一類非常值得關注的分子.實驗中在拉伸兩末端為吡啶基的金電極-分子-金電極體系并對體系電導進行測量時發現, 此類分子的拉伸電導曲線一般會出現高低電導轉換現象, 這是一般分子很少出現的比較特殊的一種現象[47?54].弄清產生這一現象的物理過程和內在物理機制, 對于利用含吡啶末端分子構建分子開關、分子存儲器、分子傳感器等功能分子器件具有十分重要的意義. 為了理解這一現象, 一些課題組通過輸運性質計算探討了體系高低電導可能對應的幾何結構. 這些探索性的設計與計算雖然在一定程度上可以說明高低電導的差異, 但未與分子結的拉伸過程相聯系, 因此這些研究結果既無法合理地理解分子結拉伸過程中體系高低電導間的突變式轉換過程, 更無法發現分子結拉伸過程中界面結構的變化以及界面原子的移動過程. 為了解決這些問題, 真正解決末端為吡啶基的分子結拉伸過程中的高低電導之謎, 發展了基于第一性原理計算的分子結絕熱拉伸模擬技術[30,37], 對 4, 4′-二吡啶分子結的拉伸過程進行了計算模擬. 計算結果不僅破解了 4, 4′-二吡啶分子與金電極構成的分子結在拉伸過程中出現的雙電導平臺之謎, 而且進一步證明了, 通過理論計算并結合相關實驗測量, 可以很好地在單原子/分子層次上有效識別電極尖端幾何結構以及分子-電極間的界面連接方式.
2 理論模型與計算過程
發現4, 4′-二吡啶分子結拉伸過程中存在高低電導轉換現象的實驗大多采用的是掃描隧道顯微裂結 (STM-BJ) 技術[50?53], 為了達到高分辨率, 掃描所用的金探針電極需要做成非常尖的錐型電極,而基底電極一般通過蒸鍍技術做成比較平整的平面, 通常基底電極會存在少量缺陷或者表面孤立金原子(如圖1所示). 另外實驗中為了方便判斷分子結的形成, 在構建分子結時, 首先控制探針電極與基底進行接觸, 然后緩慢抬起探針, 通過吸附作用將分子拉入探針與基底間隙. 這一操作過程可以使探針在與基底斷開前先產生1.0G0(G0=2e2/h為量子電導單位)的電導, 然后斷開時電導突然下降并出現分子電導信號. 考慮到實驗過程中的技術性問題以及計算的可行性, 在理論模擬中選取圖1中虛線框內的有限體系進行計算. 研究表明, 由于原子間相互作用的局域性以及次近鄰原子的屏蔽作用, 這樣選擇可以很好地模擬原子與分子間的相互作用與變化過程.
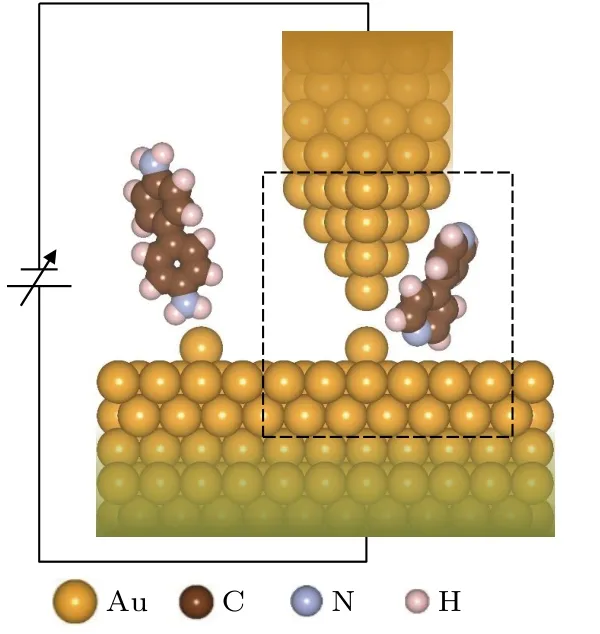
圖 1 STM-BJ 技術中分子結形成原理示意圖Fig. 1. The schematic structure of the forming principle of molecular junction in STM-BJ technique.
在分子體系拉伸過程的每一步幾何結構計算中, 兩電極的最外層金原子固定, 內層金原子及分子的坐標完全放開進行幾何結構優化, 其中兩電極最外層金原子之間的垂直距離定義為電極距離(D) (如圖2(a)所示). 分子結的拉伸是在上一步優化好的幾何結構基礎上, 將電極的最外層金原子向外移動一小段距離后固定, 然后再次對其他金原子和分子進行幾何結構優化. 這樣通過逐步結構優化, 完成基于第一性原理計算的分子結絕熱拉伸過程模擬. 體系結構優化基于雜化的密度泛函理論,計算采用B3LYP雜化交換關聯泛函, 選用LanL2DZ為基矢, 在Gaussian09程序包上進行[56].
由于實驗中測量體系電導所用的偏壓為不超過0.3 V的較低偏壓, 在這一偏壓下, 體系的共振輸運通道尚未開通, 因此體系的電導主要來源于電子的非共振輸運貢獻. 非共振輸運的特點是電子以波動的形式由一電極進入分子后, 在分子所產生勢場的散射與衍射作用下進入另一電極. 因此電流的計算采用筆者前期發展的基于分子非共振輸運機制的一維透射結合三維修正近似(OTCTCA)方法[57], 即
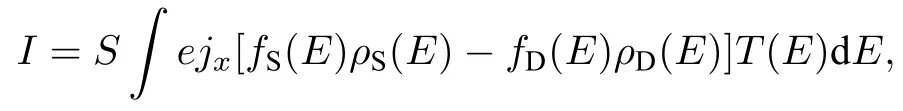
其中,jx為輸運方向的電流密度,T(E) 為透射系數,fS(E) 與fD(E) 分別為兩電子源的費米分布函數,ρS(E) 與ρD(E) 分別為兩電子源的態密度,S為電子的有效注入面積. 透射系數T(E) 依賴于體系所產生的三維有效勢場, 通過求解一維薛定諤方程, 并根據原子對電子波的衍射效應進行三維修正得到. 在以前的研究中, OTCTCA方法不僅成功計算出了分子電導與分子長度之間的指數衰減規律以及分子結拉伸過程中的電導平臺效應, 而且給出了易于理解的物理圖像[6,57].
3 計算結果與討論
3.1 4, 4′-二吡啶分子結與眾不同的拉伸過程
利用STM-BJ技術構建分子結, 探針與基底接觸時容易拉出孤立的表面金原子, 另外基底本身也會存在缺陷或者表面金原子. 計算表明吡啶環中的N原子極易吸附到基底表面的孤立金原子上,因此計算中在基底平面電極上放置了一個孤立的表面金原子, 設計了探針電極尖端為三棱錐形、基底電極含有孤立表面金原子的4, 4′-二吡啶分子結(體系 I, 如圖 2(a)—(d)所示). 同時為了比較, 計算中也設計了基底電極表面相對平整的沒有表面金原子的 4, 4′-二吡啶分子結 (體系 II, 如圖 2(e)—(g)所示). 另外, 為了與吡啶末端分子進行對比,還計算了 4, 4′-二氨基聯苯分子結的拉伸過程, 其中基底電極采用了含有孤立表面金原子的結構模式 (體系 III, 圖 2(h)—(k)).
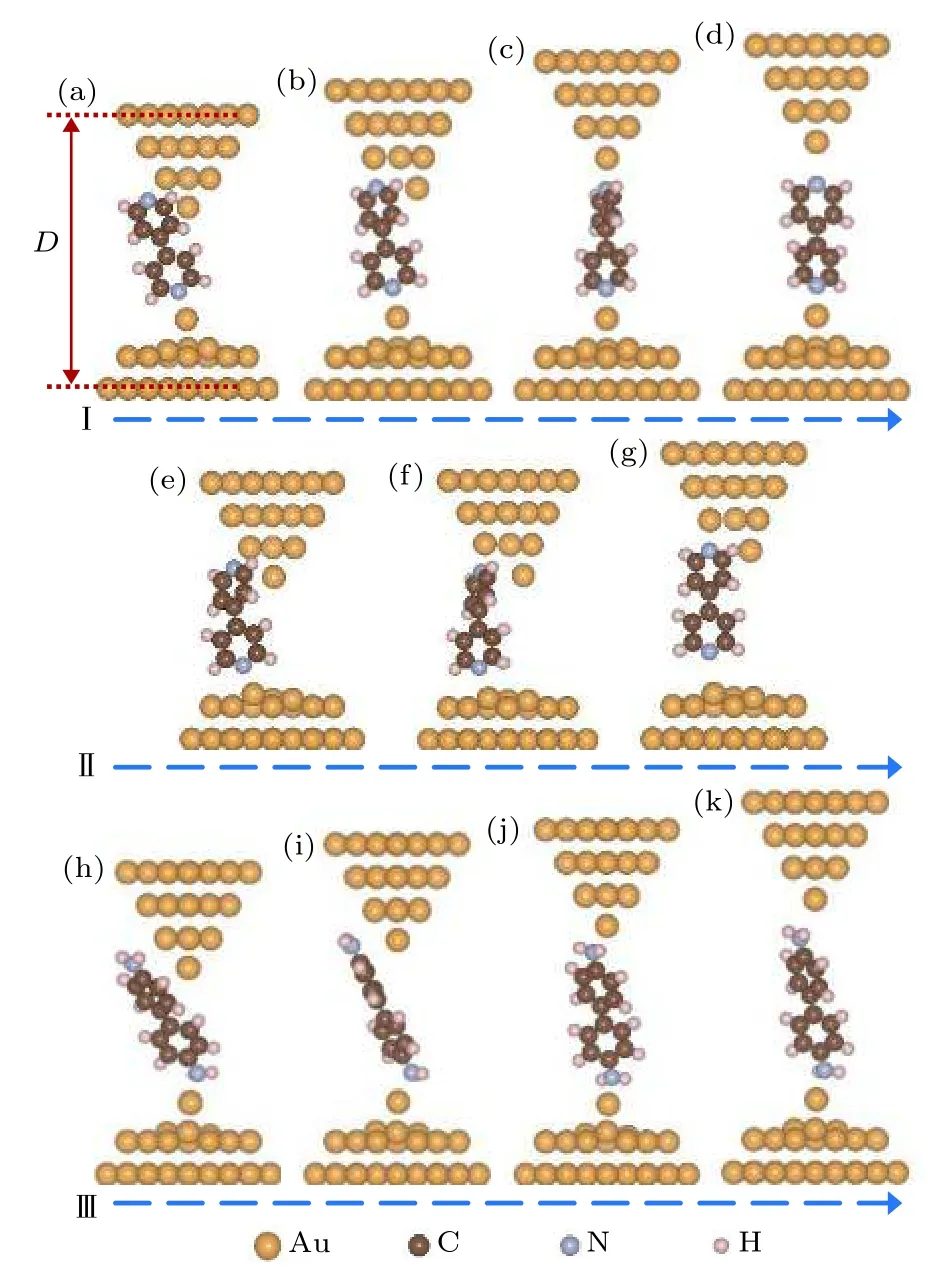
圖 2 分子體系在拉伸過程中的結構演化 (a)?(d) 吸附在基底電極表面金原子上的4, 4′-二吡啶分子結(體系I)的拉伸與結構演化過程; (e)?(g) 吸附在基底電極表面上的4, 4′-二吡啶分子結(體系II)的拉伸與結構演化過程;(h)?(k) 吸附在基底電極表面金原子上的 4, 4′-二氨基聯苯分子結(體系III)的拉伸與結構演化過程Fig. 2. Configuration evolutions in the stretching processes of molecular junctions: (a)?(d) Stretching and configuration evolution process of 4, 4′-bipyridine molecular junction,in which the 4, 4′-bipyridine molecule is adsorbed on the surface Au atom of substrate electrode (denoted as System I);(e)?(g) stretching and configuration evolution process of 4,4′-bipyridine molecular junction, in which the 4, 4′-bipyridine molecule is adsorbed on the surface of substrate electrode(denoted as System II); (h)?(k) stretching and configuration evolution process of 4, 4′-diaminobiphenyl molecular junction, in which the 4, 4′-diaminobiphenyl molecule is adsorbed on the surface Au atom of substrate electrode (denoted as System III).
圖2(a)—(d)顯示的是體系I的拉伸過程. 初始, 4, 4′-二吡啶分子末端的氮原子吸附在表面金原子上. 一般情況下, 探針探測到分子時, 分子在π電子的作用下斜靠在探針電極旁邊(圖2(a)). 隨著探針電極逐漸抬高, 由于基底電極的表面金原子對分子具有較強的吸附作用, 分子的上端在探針表面逐漸向下滑移. 當電極距離大約為2.0 nm時,分子上端的N原子開始吸附到探針尖端的第二層金原子上. 由于初始為側面吸附, 氮原子和金原子的吸附強度很弱, 其吸附作用力僅為0.1 nN左右.特別值得關注的是, 當電極距離拉伸到2.04 nm時,分子上端的氮原子與第二層金原子之間的吸附作用迅速加強, 并對尖端的金原子產生明顯的側向推動作用, 把尖端的金原子向一側推移約0.15 nm,從而使分子末端正面吸附到第二層金原子上(如圖2(b)所示). 目前根據對含各種常見末端的分子結進行第一性原理拉伸模擬計算發現, 這是含吡啶末端分子所特有的一種現象[6,8,30,37?39]. 繼續將電極距離拉伸到 2.24 nm, 4, 4′-二吡啶分子上端的氮原子從探針電極第二層金原子上斷開, 迅速連接到探針頂端的金原子上(圖2(c)), 這時頂端金原子也回到原來正常的晶格位置上. 可以發現, 4, 4′-二吡啶分子從吸附到探針電極第二層金原子到吸附到尖端金原子, 電極距離拉伸了約為 0.21 nm, 這與實驗測量結果完全一致[52?54]. 而當電極距離拉伸到 2.47 nm, 4, 4′-二吡啶分子從探針電極尖端的金原子上斷下來(圖2(d)).
圖3給出了分子體系拉伸過程中能量、作用力以及電導隨電極距離的變化曲線, 其中正作用力表示分子與電極間為吸引作用, 負作用力表示分子與電極間為擠壓作用. 圖 3(a)顯示, 當體系 I中 4, 4′-二吡啶分子上端的N原子剛吸附到第二層金原子時, 體系仍處于高能量狀態, 特別是當分子上端剛從側面吸附變為正面吸附時, 分子與電極間存在著較強的約為0.85 nN的擠壓作用. 盡管分子與電極間有較強的擠壓作用, 但是分子正面吸附到第二層金原子上具有更低的能量. 從圖中可以看出, 在2.04 nm附近分子從側面吸附到第二層金原子上轉變為正面吸附到第二層金原子上(即從圖2(a)構型轉變為圖2(b)構型)的過程中, 體系能量明顯降低了大約 0.1 eV. 另外, 從體系幾何結構的優化過程中發現, 電極距離為2.04 nm時, 探針尖端的金原子受到持續的背向氮原子的作用力, 在尖端金原子側向移動的 0.15 nm距離內, 其中大約0.1 nm的距離受力接近或超過 0.1 nN. 因此在分子結拉伸過程中, 分子可以自動地由側面吸附轉變為正面吸附. 分子正面吸附到第二層金原子上的最低能量狀態發生在2.11 nm的電極距離下. 當電極距離處于2.04—2.11 nm之間時, 分子與電極間一直保持為相互擠壓作用, 在這一過程中, 體系能量降低約 0.2 eV. 而電極距離在 2.11—2.23 nm 之間變化時, 分子與第二層金原子之間則表現為相互吸引作用, 因此在這一范圍內增大電極距離需要對電極施加一定的拉力作用, 而體系能量在這一過程中也因拉力主動做功提高了約0.5 eV. 當電極距離為 2.23—2.24 nm 時, 拉力達到約 1.2 nN, 這時拉力達到分子與第二層金原子相互作用的極限, 分子上端從第二層金原子上斷開并迅速連接到尖端金原子上, 同時體系能量迅速降低大約 0.4 eV. 當分子剛剛連接到尖端金原子上時, 分子與電極間表現出約0.8 nN的擠壓作用. 而當電極距離為2.35 nm時, 體系I達到分子與尖端金原子相連接的最低能量狀態, 與分子連接到第二層金原子上的穩定狀態相比, 體系能量降低了約 0.25 eV. 分子與尖端金原子之間能夠承受的最大拉力是0.8 nN, 對應電極距離是 2.43 nm. 可以發現, 4, 4′-二吡啶分子與探針電極第二層金原子正面連接能夠承受的最大拉力明顯高于其與尖端金原子相連接能夠承受的拉力. 這不僅與實驗觀察到的高電導狀態的斷裂力明顯高于低電導狀態的斷裂力一致, 而且與實驗測量到的斷裂力的數值也完全符合[54]. 目前技術上還無法觀察到分子結拉伸過程中分子與電極界面結構的具體變化細節, 因此基于第一性原理的絕熱拉伸計算方法不僅可以模擬分子結的拉伸過程、而且是理解并識別分子結界面特殊物理過程和分子體系特殊物理現象的非常有效的手段.
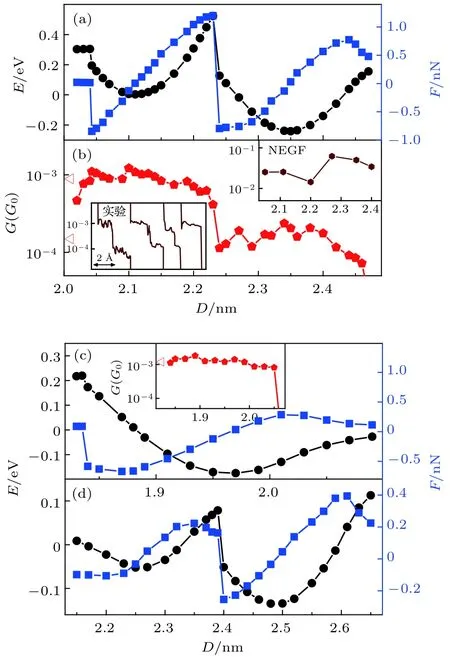
圖 3 分子體系拉伸過程中的能量、作用力和電導變化曲線 (a) 體系I拉伸過程中的能量、作用力隨電極距離的變化曲線及其(b) 電導變化曲線, 其中左下插圖為文獻[53]中實驗測量結果, 右上插圖為非平衡格林函數(NEGF)方法計算結果; (c) 體系 II 和 (d) 體系 III的能量、作用力隨電極距離的變化曲線, (c)中插圖為體系II拉伸過程中電導變化曲線Fig. 3. Energy, force, and conductance traces of the molecular junctions in the stretching processes: (a) Energy, force and (b) conductance traces as functions of electrode distances for the stretching process of system I. The bottomleft inset in (b) is the experimental conductance traces that are reported in Ref. [53], and the top-right inset in (b) is the results calculated by applying NEGF method. (c) Energy and force traces as functions of electrode distances for the stretching process of system II and (d) system III. The inset in (c) is the conductance traces of system II.
為了進一步證明在分子結拉伸過程中, 4, 4′-二吡啶分子能夠從側面吸附自動轉換成正面吸附到探針電極的第二層金原子上, 同時說明表面金原子在分子結拉伸過程中的作用, 進一步計算了基底電極無表面金原子的分子體系II的拉伸過程. 圖2(e)—(f)顯示在體系II的拉伸過程中, 當電極距離拉伸到1.84 nm時, 分子上端同樣由側面吸附變為正面吸附到探針電極的第二層金原子上, 而且探針尖端的金原子由于分子的側向推動作用移到了一側. 與體系I的拉伸過程不同的是, 當繼續增大電極距離時, 分子沒有從探針電極第二層金原子上移動到尖端金原子, 而是整個分子在第二層金原子的牽引下向上移動, 并將分子下端拉離基底電極. 由此可見,基底電極上的表面金原子不僅能夠將分子由探針電極第二層金原子上拉到尖端金原子上, 而且在整個分子結拉伸過程中保持分子不離開基底電極具有至關重要的作用. 這一點可以從分子結不同拉伸過程中作用力的差別上很明顯的表現出來. 由圖3(c)可以發現, 對于體系II, 當電極距離拉到2.1—2.3 nm, 分子逐漸被拉離基底表面時, 所需的最大拉力只有約0.3 nN, 明顯小于體系I中將分子由探針電極第二層金原子拉到尖端金原子上的1.2 nN作用力. 由此可見 4, 4′-二吡啶分子與平面金電極的吸附最大只能承受約0.3 nN的拉力作用, 而如果 4, 4′-二吡啶分子吸附到表面金原子, 則能夠承受大于1.2 nN的拉力作用. 所以表面金原子對于4, 4′-二吡啶分子具有很強的固定作用.
根據已計算的各種末端基團單分子結的拉伸過程發現, 只有含吡啶末端的分子在吸附到錐型電極第二層金原子上時才能自動由側面吸附轉換成正面吸附, 并同時產生較強的對尖端金原子的側向推動作用. 圖 2(h)—(k)是 4, 4′-二氨基聯苯分子結的拉伸過程, 從圖中可以很明顯地看出, 在分子結拉伸過程中, 上面的氨基可以很平穩地由探針電極第二層金原子滑移到尖端金原子上. 氨基末端分子結的拉伸過程及相應的電輸運特性, 在文獻[6]中已做過系統的報道.
3.2 破解4, 4′-二吡啶分子結拉伸過程中的高低電導轉換之謎
特別值得關注的是4, 4′-二吡啶分子結拉伸過程中獨特的高低電導轉換現象. 圖3(b)是基于OTCTCA方法計算得到的體系I在拉伸過程中電導隨電極距離的變化曲線. 從圖中可以看出, 4, 4′-二吡啶分子結在拉伸過程中表現出明顯的雙電導平臺現象. 其中2.02—2.22 nm之間的高電導平臺對應于4, 4′-二吡啶分子正面吸附于探針電極第二層金原子上, 而2.23—2.45 nm之間的低電導平臺對應于4, 4′-二吡啶分子吸附在探針電極的尖端金原子上. 高電導與低電導之間相差約5—8倍, 這一差別與文獻報道的實驗測量結果完全一致[50,53,54].另外, 從文獻[53]報道的實驗測量結果發現, 分子結拉伸過程中還存在只有高電導平臺、沒有低電導平臺的電導曲線(圖3(b)插圖)[53], 這與OTCTCA方法計算得到的體系II拉伸過程的電導變化曲線是一致的(圖3(c)插圖). 對于體系II, 由于基底電極上沒有表面金原子, 當分子正面吸附到第二層金原子上后, 繼續增大電極距離, 分子直接被拉離基底表面, 從而導致低電導平臺缺失. 比較體系I和體系II的拉伸過程和電導變化情況可以發現, 有兩個關鍵因素導致了4, 4′-二吡啶分子結在拉伸過程中出現高低電導轉換現象: 1)分子的吡啶末端具有較強的趨向于正面吸附到探針電極第二層金原子上的獨特性質, 并且由此產生對尖端金原子的側向推動力; 2)基底電極表面金原子對分子的吡啶基末端具有更強的吸附作用, 可以將分子由探針電極第二層金原子拉至尖端金原子上. 根據以上分析, 若 4, 4′-二吡啶分子結在拉伸過程中出現高低電導轉換現象, 則可以判斷出基底電極一定存在表面金原子, 且分子末端一定吸附在表面金原子上.這表明, 利用單分子結拉伸過程中測量到的電導曲線, 可以有效識別電極尖端構型以及分子與電極間的連接方式.
至于分子的兩種吸附狀態為什么會產生差別很大的高低電導, 可以從OTCTCA方法中得到非常好的解釋[57]. 在OTCTCA理論框架中, 電子在外加偏壓作用下由一端電極進入分子, 由于存在波動性, 電子受到分子所在空間勢場的散射后, 注入另一端電極, 從而形成電流. 從三維角度來說, 分子中的每一個原子都是電子波的一個散射中心, 由于原子核附近的勢場對于電子來說相當于勢阱, 因此可以看作電子波的小孔. 所以電子波經過每一個原子時同時發生散射和衍射, 導致的結果是電子只有一部分能夠通過原子, 而另一部分將返回源電極. 另外, 由于在電子傳播路徑上電勢是起伏變化的, 即傳播路徑上既有勢壘也有勢阱, 因此根據量子力學一維勢壘/勢阱的透射與反射理論, 電子波在傳播路徑上既有透射又有反射. 綜上可見, 電子在傳輸過程中遇到原子時, 三維效應導致電子在傳播過程中出現散射和衍射, 而一維效應導致電子在傳輸路徑上發生一定概率的反射. 在OTCTCA方法中計算電子透過率時分別從一維和三維角度考慮了分子中各原子對電子的反射、散射和衍射效應, 有效解決了低偏壓下的電子輸運問題[57].
基于OTCTCA方法可以發現, 與分子吸附到探針電極第二層金原子上相比, 4, 4′-二吡啶分子吸附到尖端金原子上時, 尖端金原子增加了對電子的散射、衍射與反射, 導致電子的透射概率降低,從而產生了低電導. 根據計算經驗, 電子在輸運路徑上每經過一個原子, 透射概率將降低大約3/4,即只有大約1/4的電子波可以繼續向前傳播. 另外分子與電極的耦合強度會影響界面電勢分布, 并在一定程度上影響透射概率. 這里需要指出的是, 在OTCTCA理論中沒有考慮擴展分子軌道進入偏壓窗而產生的共振透射的貢獻. 由于實驗是在低偏壓下測量的, 所有軌道均未開通, 即共振透射可以忽略, 因此本工作發展的OTCTCA方法不僅能給出與實驗符合很好的結果, 而且給出了容易理解的物理圖像. 對 4, 4′-二吡啶分子結的高低電導現象, 同時采用NEGF方法進行了計算, 計算基于密度泛函理論, 采用廣義梯度近似(GGA)下的PBE(Perdew-Burke-Ernzerhof)泛函、截止能 (mesh cut-off energy)為 200 Ry,k點取樣為 (5, 5, 100),總能量收斂標準為 1 0?6, Au原子采用單z極化(SZP)原子軌道基函數, 其余原子則采用雙z極化(DZP)原子軌道基函數, 計算在ATK軟件包[58?60]中進行. 但出人意料的是, 其計算結果不僅比實驗結果高出1個甚至2個數量級, 而且與實驗測量結果完全相反, 即分子與探針電極第二層金原子相連的較短電極距離下, 體系為低電導狀態, 而當分子與探針電極尖端金原子相連時表現為高電導(如圖3(b)插圖所示). 根據兩種理論框架的差異分析產生這一結果的內在原因發現, 與OTCTCA方法相比, NEGF在計算分子器件電流時考慮的是擴展軌道的共振輸運(即透射譜共振峰的貢獻)和偏離共振輸運(即共振峰尾部的貢獻), 而沒有考慮與共振通道完全無關的非共振輸運, 因此NEGF方法對于主要因原子對電子的散射、衍射以及反射等效應帶來的非共振輸運貢獻難以準確地反映體系的電輸運性質, 從而導致其計算結果完全偏離實驗測量結果甚至與實驗結果相反.
3.3 影響體系高低電導差異的其他因素
除了探針電極尖端金原子的散射、衍射和反射作用影響體系高低電導的差異外, 分子與電極之間軌道耦合程度的不同也在一定程度上加大了體系高低電導之間的差異. 這一點可以從擴展分子軌道的多少上很直觀地發現體系高低電導狀態之間的差異. 圖4給出了同時擴展到探針電極和分子上的所有分子軌道的空間分布圖. 從圖中可以看出, 當分子正面連接到第二層金原子上時, 有16條分子軌道同時擴展到探針電極和分子上(圖4體系I(b)所示軌道); 而當分子移動到電極尖端金原子上時, 只有8條分子軌道同時擴展到探針電極和分子上(圖4體系I (c)所示軌道). 擴展分子軌道數目的差異表明, 當分子正面吸附到第二層金原子時,分子與電極間具有更好的耦合作用. 因此盡管低偏壓下這些軌道離偏壓窗很遠, 但分子與電極間較強的軌道耦合仍然可以有效提高電子在分子與電極間非共振轉移的概率. 這是因為這些軌道可以通過影響分子與電極界面處電勢的空間分布來影響電子的輸運性質.
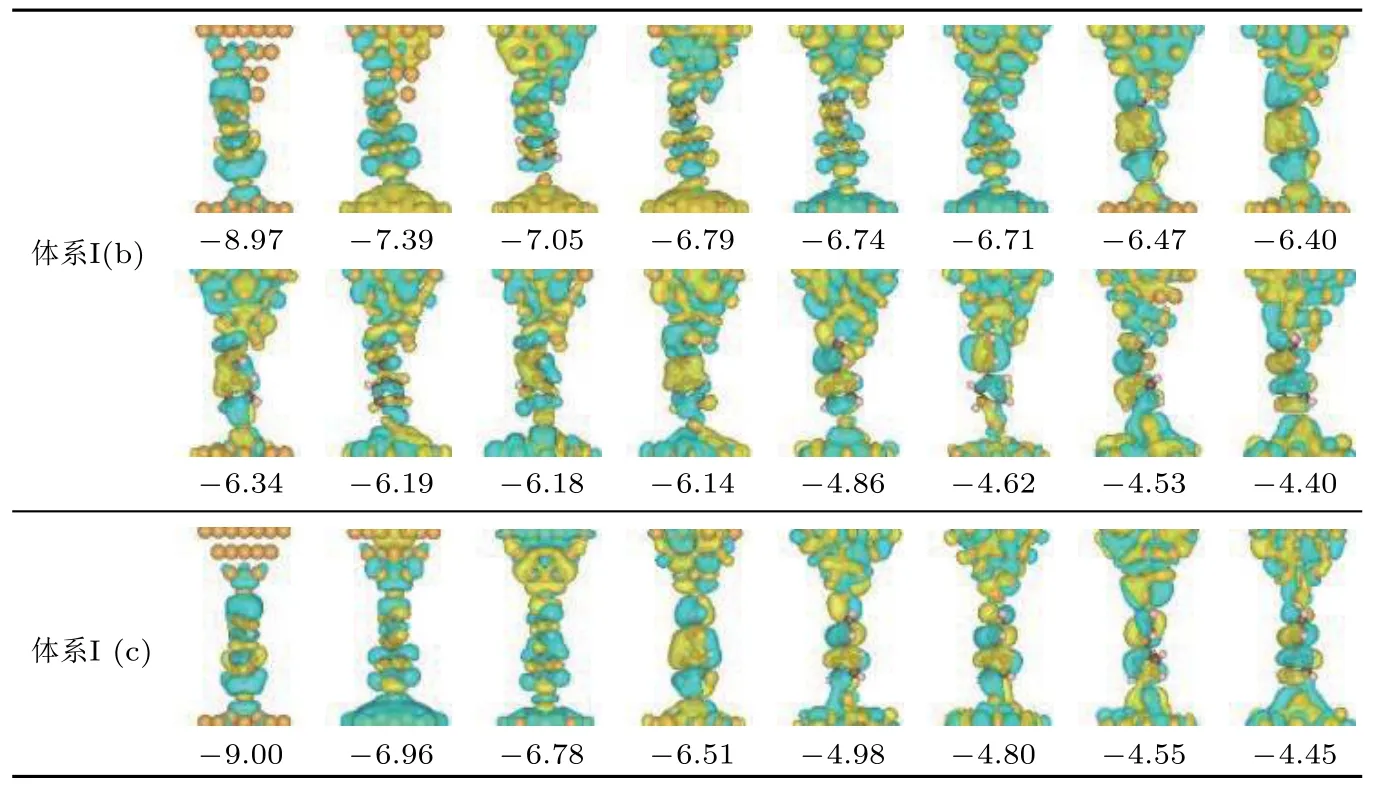
圖 4 圖2體系I (b)和體系I (c)中同時擴展到分子與探針電極上的所有占據分子軌道空間分布圖, 圖中數字為各軌道相對于費米能級的能量(單位: eV)Fig. 4. Spatial distributions of occupied molecular orbitals of System I (b) and System I (c) in Fig. 2 that are delocalized on the molecule and probe electrode simultaneously. The numbers in the figures are the orbital energy relative to the Fermi level (the unit is eV).
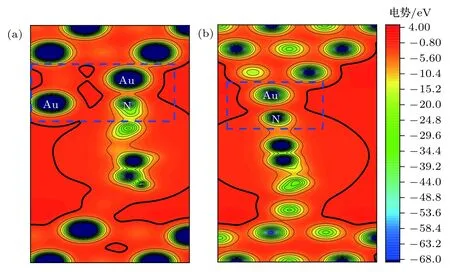
圖 5 (a) 分子吸附到探針電極第二層金原子上 (圖 2中體系 I (b))和 (b) 分子吸附到探針電極尖端金原子上 (圖 2中體系 I (c))體系所在的空間的電勢分布圖Fig. 5. (a) Spatial distributions of potential of the system that the molecule adsorbs on the second gold layer of prob electrode(system I (b) in Fig. 2) and (b) the system that the molecule adsorbs on the top gold of prob electrode (system I (c) in Fig. 2).
圖5對分子吸附到探針電極第二層金原子上和分子吸附到探針電極尖端金原子上體系所在空間的電勢分布進行了比較, 圖中的藍色虛線框內是分子與探針電極連接區域的勢阱通道. 從圖中可以看出分子連接到第二層金原子上后, 由于分子與電極的耦合增強, 電子在電極與分子之間轉移的勢阱通道(黑色實線內側區域)明顯比分子連接到電極尖端金原子上的勢阱通道寬. 根據量子力學理論和OTCTCA計算, 電子在勢壘中是以衰減波的形式傳輸, 即電子的透射概率隨著勢壘在輸運方向長度的增大而迅速衰減; 而電子在勢阱中則是以平面波或者球面波的形式傳播, 傳播過程中會因勢阱的起伏發生一定的反射, 但不會因勢阱在輸運方向長度的增大而衰減. 因此在低偏壓沒有共振輸運通道開通的情況下, 電子通過勢阱通道的非共振傳輸便成為體系電流的主要來源. 因此與分子連接到探針電極的尖端金原子上相比, 分子連接到探針第二層金原子上拓寬了電子在分子與電極間轉移的勢阱通道, 所以必然提高電子在分子與電極間的轉移概率和體系的導電能力. 另外從體系結構上還可以發現, 4, 4′-二吡啶分子正面連接到第二層金原子后,有4個金原子與分子近鄰, 且電極距離更短, 而分子連接到尖端金原子上時, 與分子近鄰的金原子只有1個, 這也在一定程度上影響著電子的透射概率.
4 結 論
基于第一性原理絕熱拉伸模擬方法研究了4, 4′-二吡啶分子結拉伸過程中出現的高低電導轉換現象. 結果顯示當 4, 4′-二吡啶分子吸附到探針電極的第二層金原子上后, 吡啶基末端產生了特有的側向推動力, 將探針尖端金原子推向一側, 使得分子由側面吸附到第二層金原子上轉變為正面吸附到第二層金原子, 從而導致分子結出現高電導狀態. 進一步將電極距離拉伸約0.22 nm后, 分子末端的氮原子由探針電極第二層金原子滑移至尖端金原子上, 分子結呈現低電導狀態. 另外, 計算結果進一步顯示, 基底電極表面存在孤立金原子, 且只有分子吸附在基底表面孤立金原子上, 分子結才會在拉伸過程中出現高低電導轉換現象. 若分子未吸附在基底表面孤立金原子上, 則不能將分子從探針電極第二層金原子上拉至尖端金原子上, 從而導致低電導狀態缺失. 本工作的計算不僅破解了4, 4′-二吡啶分子結拉伸過程中的高低電導之謎, 同時提供了一種在原子尺度上識別界面結構和理解體系物理過程的有效理論方法.
猜你喜歡
商品與質量(2021年43期)2022-01-18 05:31:22
杭州(2020年23期)2021-01-11 00:54:42
新世紀智能(數學備考)(2020年11期)2021-01-04 00:38:16
中國外匯(2019年17期)2019-11-16 09:31:14
中國衛生(2015年12期)2015-11-10 05:13:40
現代企業(2015年1期)2015-02-28 18:43:18
汽車零部件(2014年5期)2014-11-11 12:24:28
新高考·高一物理(2014年1期)2014-09-18 01:26:07
浙江人大(2014年1期)2014-03-20 16:19:53
終身教育研究(2012年4期)2012-03-25 10:41:11
