HPLC 法同時測定甘草中3 種成分
2020-11-02 13:14:46靳雪飛何云嬌施務(wù)務(wù)葛少波褚瑞萌
中成藥 2020年10期
關(guān)鍵詞:方法
靳雪飛 何云嬌 張 杰 施務(wù)務(wù) 葛少波 褚瑞萌
(1.蚌埠市第一人民醫(yī)院中藥房, 安徽 蚌埠233000; 2.安徽中醫(yī)藥大學第一附屬醫(yī)院藥學部, 安徽 合肥230009)
甘草是臨床常用中藥,具有補脾益氣、清熱解毒、祛痰止咳、緩急止痛、調(diào)和諸藥的作用,被譽為“眾藥之王”,甘草苷、甘草酸、甘草次酸是其主要活性成分[1?2],均為黃酮類化合物,具有解毒、抗炎、抗病毒、抗腫瘤等作用,其中甘草苷、甘草酸具有增加免疫、保護阿爾茨海默腦、抗氧化、抗糖尿病、抗哮喘等活性,而甘草次酸為相對高效的抗癌成分,具有消炎、抗菌、抗感染、抗病毒、抗HIV、增強機體對毒物耐受等。
現(xiàn)有報道主要集中于對甘草有效部位提取分離的研究,以及對臨床療效的探討,而對其活性成分準確測定的報道相對較少,并且也只針對其中一兩個化合物,尚無同時對甘草苷、甘草酸、甘草次酸的有效快速定量檢測。因此,本實驗建立HPLC 法同時測定甘草中上述3 種成分的含有量,以期為市場上對該藥材質(zhì)量控制提供依據(jù),也對相應制劑的制備具有重要意義[3?5]。
1 材料
HP?1100 高效液相色譜儀(北京科瑞邁科技有限責任公司)。甘草購自東營市益生堂醫(yī)藥連鎖有限公司(批號160201、170401、170602),經(jīng)專家鑒定為正品。甘草酸銨、甘草苷、甘草次酸對照品均由中國食品藥品檢定研究院提供,批號分別為 110731?200407、111610?201005、110723?201715。甲 醇(批 號 17065013)、乙 腈(批 號174488)為色譜純(美國賽默飛世爾科技公司);醋酸銨(批號171102)為分析純(廣州市三昌化工有限公司)。
2 方法與結(jié)果
2.1 色譜條件 Kromasil C18色譜柱(4.6 mm×250 mm,5 μm);流動相乙腈(A)?0.03 mmol/L 醋酸銨(B),梯度洗脫,程序見表1;體積流量1.0 mL/min;柱溫28 ℃;檢測波長327 μm(0~5 min)、237 nm(5~20 min);進樣量20 μL。色譜圖見圖1。

表1 梯度洗脫程序
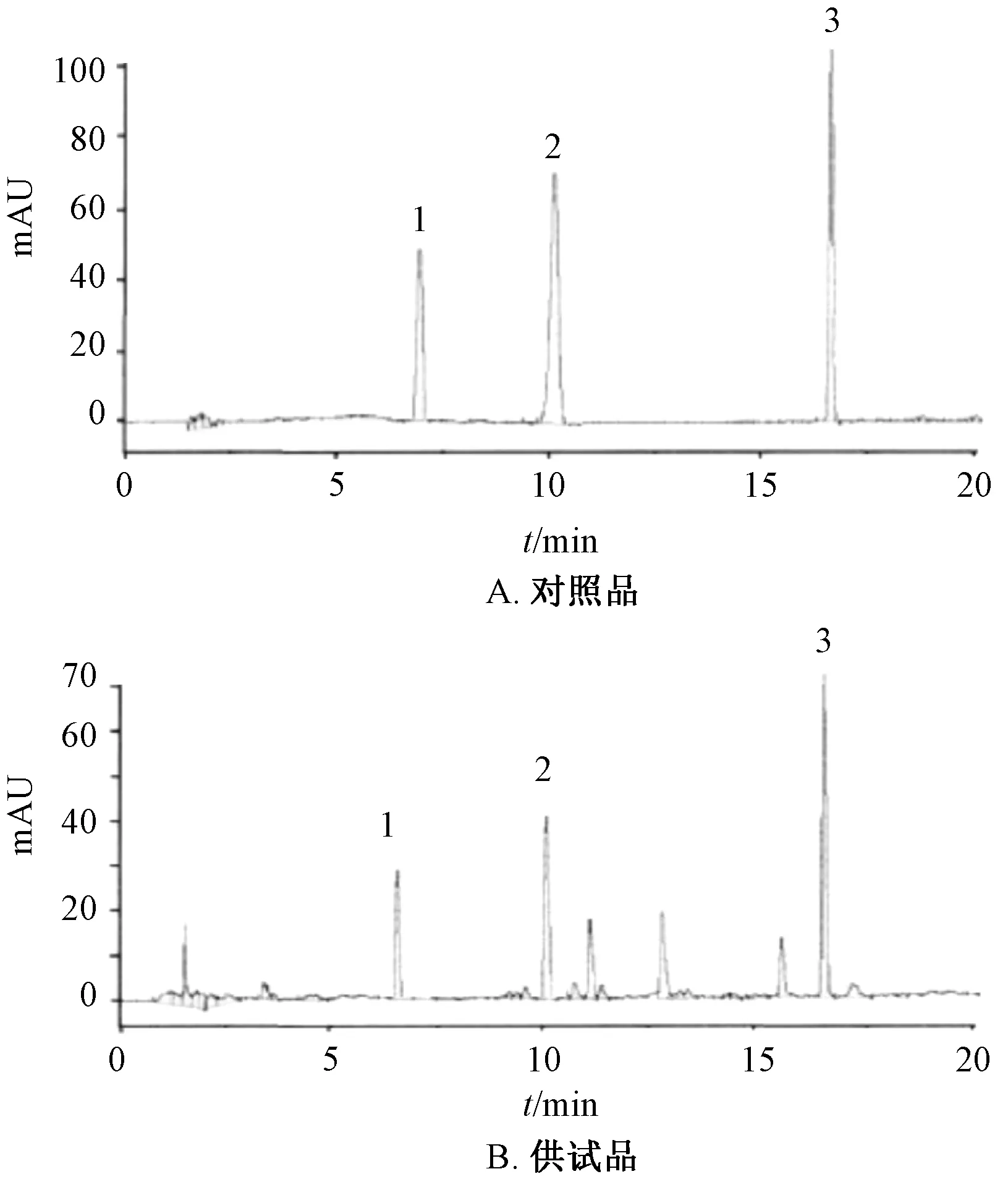
圖1 各成分HPLC 色譜圖
2.2 溶液制備
2.2.1 供試品溶液 精密稱取飲片0.2 g,置于100 mL 圓形燒瓶中,加入50%甲醇100 mL,回流提取2 h 后在70 ℃下回收溶劑,放冷,稱定質(zhì)量,50%甲醇補足減失的質(zhì)量,混勻,0.45 μm 微孔濾膜過濾,取續(xù)濾液,即得。
2.2.2 對照品溶液 精密稱取甘草酸胺、甘草苷、甘草次酸對照品適量,50%甲醇溶解稀釋至刻度,即得,每1 mL分別含三者0.04、0.02、0.08 mg(甘草酸含有量=甘草酸胺含有量/1.020 7)。
2.3 線性關(guān)系考察 精密吸取對照品溶液5、10、15、20、25、30 μL,在“2.1” 項色譜條件下進樣測定。以進樣量為橫坐標(X),峰面積為縱坐標(Y)進行回歸,得方程分別為甘草酸Y=25.841X-14.26(r=0.998 0)、甘草苷Y=22.197X+10.374(r=0.999 0)、甘草次酸Y=18.722X-10.928(r=0.999 0),分別在0.201~1.206、0.113~0.678、0.392~2.352 μg 范圍內(nèi)線性關(guān)系均良好。
2.4 方法學考察
2.4.1 精密度試驗 取對照品溶液20 μL,在“2.1” 項色譜條件下進樣測定6 次,測得甘草酸胺、甘草苷、甘草次酸峰面積RSD 分別為0.18%、0.42%、0.87%,表明儀器精密度良好。
2.4.2 穩(wěn)定性試驗 取同一份供試品溶液,于0、2、4、6、8、10 h 在“2.1” 項色譜條件下進樣測定,測得甘草酸胺、甘草苷、甘草次酸峰面積RSD 分別為0.91%、1.04%、1.48%,表明溶液在10 h 穩(wěn)定性良好。
2.4.3 重復性試驗 取同一批飲片,按“2.2.1” 項下方法平行制備6 份供試品溶液,測得甘草酸胺、甘草苷、甘草次酸峰面積RSD 分別為1.38%、1.49%、1.45%,表明該方法重復性良好。
2.4.4 加樣回收率試驗 精密稱取各成分含有量已知的飲片6 份,每份0.2 g,置于100 mL 圓形燒瓶中,加入1 mL對照品溶液,按“2.2.1” 項下方法制備供試品溶液,在“2.1” 項色譜條件下進樣測定,計算回收率。結(jié)果,甘草酸、甘草苷、甘草次酸平均加樣回收率分別為98.33%、99.43%、98.47%,RSD 分別為0.89%、0.47%、0.65%。
2.4.5 樣品含有量測定 取3 批飲片,每批6 份,按“2.2.1” 項下方法制備供試品溶液,在“2.1” 項色譜條件下進樣測定,計算含有量,結(jié)果見表2,可知甘草苷、甘草酸含有量均符合2015 年版《中國藥典》 規(guī)定(分別不低于0.5%、2.0%)。

表2 各成分含有量測定結(jié)果(n=6)
3 討論
3.1 供試品溶液制備方法篩選 本實驗考察了50%甲醇、甲醇、50%乙醇,以及加熱回流1、2、3 h 對各成分提取效果的影響,發(fā)現(xiàn)50%甲醇加熱回流2 h 時各成分提取較完全,而且雜質(zhì)峰較少,故選擇其作為供試品溶液制備方法。
3.2 色譜條件篩選
3.2.1 色譜柱 本實驗考察了3 種不同廠家的Kromasil C18、YMC?ODS、Agilent SB?C18色譜柱(250 mm×4.6 mm,5 μm),結(jié)合方法耐用性、分離度、理論塔板數(shù)、綜合對稱因子等因素,最終選擇Kromasil C18色譜柱。
3.2.2 流動相 本實驗以乙腈替代甲醇,趙建軍等[6]報道它具有更好的洗脫能力。再通過查閱關(guān)于甘草酸、甘草苷含有量測定的文獻,選擇乙腈?0.03 mmol/L 醋酸銨作為流動相,同時不斷優(yōu)化其比例,發(fā)現(xiàn)按表1 程序梯度洗脫時分離效果好,干擾小,出峰時間短[7]。
3.2.3 檢測波長 參照2015 年版《中國藥典》,本實驗分別以327、237 nm 為第1、第2 檢測波長,可提高檢測的靈敏度和分析效率。
3.3 指標成分篩選 甘草為豆科植物,被廣泛用于抗炎、抗心血管疾病、抗病毒[8?9],其所含甘草酸是重要成分,為無色柱狀結(jié)晶體;甘草次酸是甘草酸水解成分,具有降壓、抗炎、增強機體免疫功能作用,還作為五環(huán)三萜化合物制劑的原料用于抗腫瘤[10];甘草苷為二氫黃酮類化合物,具有抗氧化、清除自由基作用,還能保護心肌細胞。因此,本實驗選擇上述3 種成分作為指標進行檢測。
4 結(jié)論
既往關(guān)于甘草的研究大多從單一成分出發(fā),缺少對主要化合物的聯(lián)合考察[11?12],可能會影響其所含多種有效物質(zhì)的提取效果。因此,本實驗建立HPLC 法同時測定甘草中甘草苷、甘草酸、甘草次酸的含有量,該方法快速準確,重復性、穩(wěn)定性良好,有助于改進該藥材提取工藝,也可為其質(zhì)量控制提供參考。
猜你喜歡
中老年保健(2021年9期)2021-08-24 03:52:04
河北畫報(2021年2期)2021-05-25 02:07:46
中學生數(shù)理化(高中版.高考理化)(2020年2期)2020-04-21 05:33:04
兒童繪本(2020年5期)2020-04-07 17:46:30
兒童故事畫報(2019年5期)2019-05-26 14:26:14
Coco薇(2016年2期)2016-03-22 02:42:52
山東青年(2016年1期)2016-02-28 14:25:23
Coco薇(2015年1期)2015-08-13 02:47:34
小雪花·成長指南(2015年7期)2015-08-11 15:03:12
小雪花·成長指南(2015年4期)2015-05-19 14:47:56
