王不留行炮制前后的UPLC指紋圖譜比較及刺桐堿和王不留行黃酮苷的含量測定
2020-10-30 01:55:26曹斯瓊吳文平羅宇琴馬瑞瑞潘禮業李國衛陳向東
中國藥房 2020年19期
關鍵詞:黃酮
曹斯瓊 吳文平 羅宇琴 馬瑞瑞 潘禮業 李國衛 陳向東

摘 要 目的:比較王不留行炮制(清炒)前后的指紋圖譜差異,并測定其炒制前后刺桐堿、王不留行黃酮苷的含量。方法:采用超高效液相色譜(UPLC)法,色譜柱為YMC Trait C18,流動相為乙腈-水(梯度洗脫),流速為0.35 mL/min,檢測波長為219 nm,柱溫為35 ℃,進樣量為1 μL。以王不留行黃酮苷為參照,繪制王不留行生品及其炮制品(各17批,編號分別為S1~S17、CS18~CS34)的指紋圖譜;采用《中藥色譜指紋圖譜相似度評價系統(2012版)》進行相似度評價及共有峰指認;采用SPSS 20.0軟件進行聚類分析、主成分分析和因子分析。采用上述UPLC法測定王不留行生品及其炮制品中刺桐堿、王不留行黃酮苷的含量。結果:17批王不留行生品及其炮制品的UPLC指紋圖譜中均有共有峰5個,相似度均大于0.99;共指認了刺桐堿和王不留行黃酮苷等2個共有峰。聚類分析結果顯示,S1~S17聚為一類,CS18~CS34聚為一類;主成分分析及因子分析結果顯示,第1主成分的方差貢獻率為76.418%,刺桐堿、王不留行黃酮苷在第1主成分上有較高載荷(特征值分別為0.976、0.966)。刺桐堿、王不留行黃酮苷檢測質量濃度的線性范圍分別為6.437~321.832、7.729~386.437 μg/mL(r均大于0.999);檢測限、定量限分別為0.085、0.284 ng(生品)和0.739、2.465 ng(炮制品);精密度、重復性、穩定性(12 h)、耐用性試驗的RSD均小于3%(n=6或n=5);平均加樣回收率分別為96.42%(RSD=0.85%,n=6)、99.13%(RSD=1.74%,n=6)。兩種成分的含量分別為0.11%~0.20%、0.42%~0.63%(生品)和0.08%~0.11%、0.34%~0.50%(炮制品)。結論:成功建立了王不留行生品及其炮制品的UPLC指紋圖譜。王不留行炒制前后的化學成分雖一致性較好,但炒制后刺桐堿、王不留行黃酮苷的含量均有所降低。
關鍵詞 王不留行;炮制;超高效液相色譜法;指紋圖譜;刺桐堿;王不留行黃酮苷;聚類分析;主成分分析
ABSTRACT ? OBJECTIVE: Compare the fingerprint difference of Vaccariae Semen before and after processed (stir-fried), and to determine the contents of erythrine and vaccarin before and after stir-fried. METHODS: UPLC method was adopted. The determination was performed on YMC Trait C18 column with mobile phase consisted of acetonitrile-water (gradient elution) at the flow rate of 0.35 mL/min. The detection wavelength was set at 219 nm, and the column temperature was 35 ℃. The sample size was 1 μL. Using vaccarin as reference, the fingerprints of Vaccariae Semen crude product and its processed product (each of 17 batches, S1-S17, CS18-CS34) were drawn. The similarity evaluation and common peak identification were carried out by Similarity Evaluation System of TCM Chromatographic Fingerprint (2012 edition); cluster analysis, principle component analysis (PCA) and factor analysis were performed by using SPSS 20.0 software. The contents of erythrine and vaccarin in Vaccariae Semen crude product and its processed product were determined by UPLC. RESULTS: There were 5 common peaks in UPLC fingerprints of 17 batches of Vaccariae Semen crude product and its processed product. The similarities were all higher than 0.99. Among them, 2 common peaks were identified, i.e. erythrine, vaccarin. Results of cluster analysis showed that S1-S17 were clustered into one category and CS18-CS34 were clustered into one category. Results of PCA and factor analysis showed that variance contribution rate of the first principle component was 76.418%; erythrine and vaccarin had higher loading on the first principal component (eigenvalues were 0.976 and 0.966, respectively). The linear ranges of above 2 components were 6.437-321.832 μg/mL and 7.729-386.437 μg/mL,respectively (r>0.999). The limits of detection and quantitation were 0.085,0.284 ng (crude product) and 0.739, 2.465 ng (processed product), respectively. RSDs of precision,reproducibility, stability (12 h) and durability tests were all lower than 3%(n=6 or n=5). Average recoveries were 96.42% (RSD=0.85%, n=6) and 99.13% (RSD=1.74%, n=6). The contents of the two components were 0.11%-0.20%, 0.42%-0.63%(crude product)and 0.08%-0.11%, 0.34%-0.50%(processed product). CONCLUSIONS: UPLC fingerprint of Vaccariae Semen crude product and its processed product are established successfully. Although the chemical constituents in Vaccariae Semen are consistent before and after stir-fried, the contents of erythrine and vaccarin are all decreased after stir-fried.
KEYWORDS ? Vaccariae Semen; Processing; UPLC; Fingerprint; Erythrine; Vaccarin; Cluster analysis; Principal component analysis
王不留行為石竹科植物麥藍菜Vaccaria segetalis (Neck.) Garcke的干燥成熟種子,具有活血化瘀、下乳消腫、利尿通淋之功效[1]。該藥味苦、性平,歸肝、胃經,可用于治療乳腺癌、產后缺乳、泌尿系統疾病等[2]。據文獻報道,該藥的主要化學成分包括黃酮苷類、生物堿類、三萜皂苷類、環肽類和脂肪油類等[3-5]。其中,黃酮苷類成分是王不留行催乳的有效成分,具有促進乳汁分泌的作用[6];而且,該類成分還能保護血管內皮細胞,促進內皮細胞增殖[7-8];此外,王不留行中的刺桐堿具有抗炎、增強免疫和抗肝損傷的作用[9]。
中藥自古有“逢子必炒”的說法,種子類中藥炒制后,種皮爆裂,質地酥脆,有利于有效成分溶出,也有助于生物利用度的提高[10-11]。2015年版《中國藥典》(一部)收載的王不留行的炮制方法為清炒法[1]。據文獻研究報道,王不留行炮制前后脂溶性成分含量及浸出物含量有明顯差異[12];炮制對王不留行中環肽A、B、E含量的影響較小[13],但炮制后其黃酮苷的含量大幅度下降,而異牡荊素-2″-O-阿拉伯糖苷的含量卻大幅上升[14-15]。指紋圖譜作為一種體現中藥化學成分整體特征的質量評價方法,對藥材的全面控制具有重要作用。本研究通過建立王不留行生品及其炮制品的超高效液相色譜(UPLC)指紋圖譜,應用化學模式識別方法比較兩者的差異性,并測定兩者中刺桐堿和王不留行黃酮苷的含量,以期為王不留行生品及其炮制品的質量控制及綜合評價提供參考。
1 材料
1.1 儀器
H-Class型UPLC儀(美國Waters公司);XP26型百萬分之一分析天平、ME204E型萬分之一分析天平(瑞士Mettler Toledo公司);KQ-500DE型數控超聲波清洗器(昆山市超聲儀器有限公司)。
1.2 藥品與試劑
王不留行黃酮苷對照品(中國食品藥品檢定研究院,批號:111853-201704,純度:99.7%);刺桐堿對照品(上海源葉生物科技有限公司,批號:P26N6F6550,純度:98.0%);乙腈為色譜純,其余試劑均為分析純,水為超純水。17批王不留行藥材購自河北、河南、安徽,經廣東一方制藥有限公司魏梅主任中藥師鑒定,均為石竹科植物麥藍菜V. segetalis (Neck.) Garcke的干燥成熟種子。17批藥材來源信息見表1。
2 方法與結果
2.1 樣品炮制
取王不留行藥材,按2015年版《中國藥典》(一部)王不留行項下“王不留行”炮制方法[1],除去雜質,得王不留行生品飲片(編號:S1~S17);按“炒王不留行”炮制方法,取凈王不留行,照“清炒法”炒至大多爆開白花,得炒王不留行飲片(編號:CS18~CS34)。
2.2 混合對照品溶液制備
精密稱取刺桐堿對照品3.344 mg、王不留行黃酮苷對照品3.011 mg,加甲醇制成每1 mL含刺桐堿32.771 ? μg、王不留行黃酮苷30.020 μg的混合對照品溶液。
2.3 供試品溶液制備
取飲片樣品,粉粹,過三號篩,取粉末約1 g,精密稱定,置具塞錐形瓶中,精密加入75%乙醇25 mL,稱定質量,超聲(功率:250 W,頻率:40 kHz)處理30 min,靜置至室溫,再稱定質量,用75%乙醇補足減失的質量,搖勻,濾過,取續濾液,即得。
2.4 色譜條件
色譜柱:YMC Trait C18(100 mm×2.1 mm,1.9 μm);流動相:乙腈(A)-水(B),梯度洗脫(0~5.5 min,10%A→15%A;5.5~11 min,15%A→30%A;11~16.5 min,30%A→70% A);流速:0.35 mL/min;柱溫:35 ℃;檢測波長:219 nm;進樣量:1 μL。
2.5 UPLC指紋圖譜的建立
2.5.1 精密度試驗 取“2.3”項下同一供試品溶液(編號:S3)適量,按“2.4”項下色譜條件連續進樣測定6次,記錄色譜圖。以王不留行黃酮苷峰為參照,計算各共有峰的相對保留時間和相對峰面積。結果,各共有峰相對保留時間和相對峰面積的RSD均小于1.0%(n=6),表明本方法精密度良好。
2.5.2 穩定性試驗 取“2.3”項下同一供試品溶液(編號:S3)適量,分別于室溫下放置0、2、4、6、12 h時按“2.4”項下色譜條件進樣測定,記錄色譜圖。以王不留行黃酮苷峰為參照,計算各共有峰的相對保留時間和相對峰面積。結果,各共有峰相對保留時間和相對峰面積的RSD均小于3.0%(n=5),表明供試品溶液于室溫下放置12 h內穩定性良好。
2.5.3 重復性試驗 取飲片樣品(編號:S3)粉碎,過三號篩,取粉末適量,共6份,分別按“2.3”項下方法制備供試品溶液,再按“2.4”項下色譜條件進樣測定,記錄色譜圖。以王不留行黃酮苷峰為參照,計算各共有峰的相對保留時間和相對峰面積。結果,各共有峰相對保留時間和相對峰面積的RSD均小于1.0%(n=6),表明本方法重復性良好。
2.6 UPLC指紋圖譜的生成及相似度評價
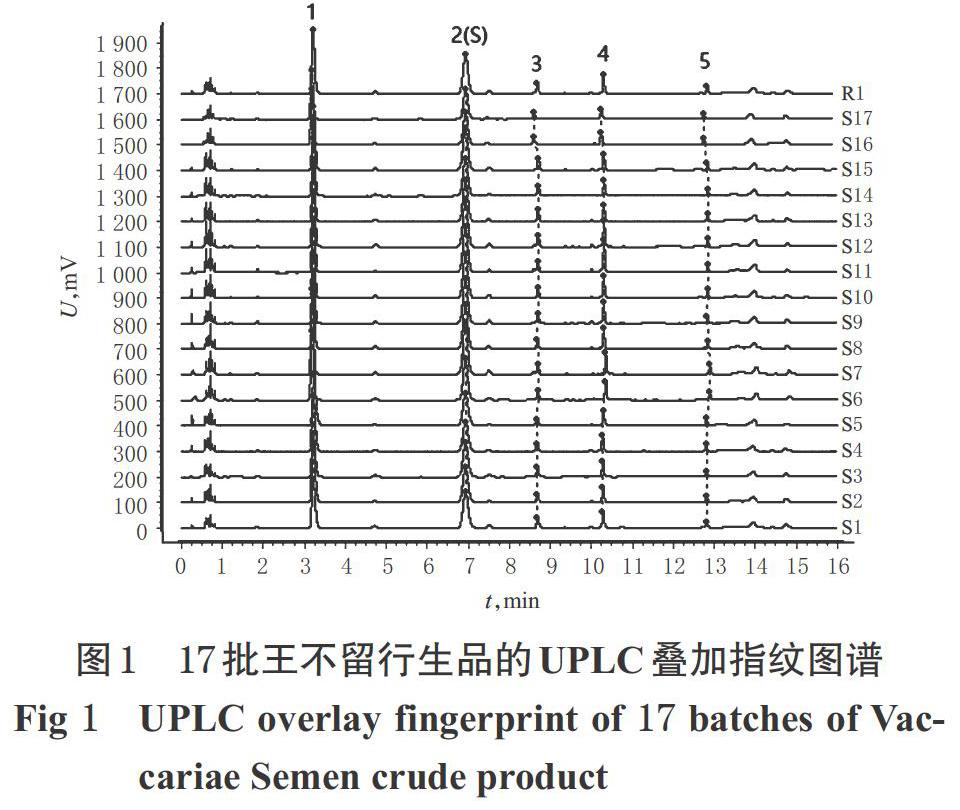
分別取17批王不留行生品飲片和17批炒王不留行飲片,按“2.3”項下方法制備供試品溶液,再按“2.4”項下色譜條件進樣測定,記錄色譜圖。將圖譜導入《中藥色譜指紋圖譜相似度評價系統(2012版)》,分別以S1、CS18樣品的指紋圖譜為參照圖譜,以2號峰為參照峰(S峰)進行保留時間校正并進行全峰匹配,生成疊加指紋圖譜,采用平均數法生成共有模式圖(對照特征圖譜為R1、R2),詳見圖1、圖2。
由圖1、圖2可見,各共有峰較穩定,具有指紋圖譜特征性,可初步認定為王不留行生品與其炮制品的指標成分群。王不留行生品和炒王不留行的UPLC疊加指紋圖譜中,分別有共有峰5個;通過與混合對照品溶液進行比對,指認了其中2個成分,即1號色譜峰為刺桐堿峰,2號色譜峰為王不留行黃酮苷峰。因王不留行黃酮苷峰分離效果好、出峰穩定且保留時間適中,故以其保留時間和峰面積作為參照(S)。共有峰對照品歸屬色譜圖見圖3。
以共有模式作為對照圖譜,將17批王不留行和17批炒王不留行分別進行相似度評價。結果,所有批次樣品的相似度均大于0.99,表明王不留行生品與其炮制品的化學成分一致性較好,詳見表2。
2.7 指紋圖譜的化學計量學分析
2.7.1 聚類分析 以各共有峰的峰面積為原始數據,運用SPSS 20.0軟件,采用組間平均數聯結法,以夾角余弦作為樣品相似度的距離公式,對17批王不留行生品和炒王不留行進行系統聚類。結果,S1~S17王不留行生品樣品聚為一類,CS18~CS34炒王不留行樣品聚為一類,提示清炒這一炮制方法對王不留行的化學成分含量有一定的影響,詳見圖4。
2.7.2 主成分分析及因子分析 以各共有峰的峰面積為原始數據,利用SPSS 20.0軟件對17批王不留行生品和17批炒王不留行指紋圖譜所得的5個共有峰進行主成分分析,得到相關矩陣的特征值及其方差貢獻率,詳見表3;得到初始因子載荷矩陣,詳見表4。提取表3中特征值>1的成分(主成分),僅第1成分符合要求,計算得其方差貢獻率為76.418%,表明該主成分代表了王不留行生品和炒王不留行中5個成分76.418%的信息量,具有很好的代表性。由表4可知,峰1和峰2在第1主成分上有較高載荷,峰1(刺桐堿)的特征值為0.976,峰2(王不留行黃酮苷)的特征值為0.966,說明第1主成分主要反映了刺酮堿和王不留行黃酮苷的信息,是用以區分王不留行生品和炒王不留行的重要因素。第1主成分可代表王不留行與炒王不留行指紋圖譜的大部分信息,主成分分析結果基本顯示出了不同批次王不留行樣品之間的相似度和差異性。按方差貢獻率計算各樣品第1主成分得分,結果顯示,王不留行生品得分較炒王不留行高,第1主成分可作為區分兩種樣品的指標,詳見表5。根據主成分分析結果繪制散點圖,詳見圖5。由圖5可知,本試驗中的樣品可以分為2類,即王不留行生品(S1~S17)為一類,炒王不留行(CS18~CS34)為一類。
2.8 刺桐堿和王不留行黃酮苷的含量測定
由“2.7”項下可知,峰1和峰2對區分王不留行生品和炒王不留行的指紋圖譜起著決定性作用,故本研究采用UPLC法對刺桐堿和王不留行黃酮苷2種成分的含量進行測定。
2.8.1 色譜條件 同“2.4”項下色譜條件。
2.8.2 系統適用性試驗 取“2.2”項下混合對照品溶液、“2.3”項下供試品溶液和陰性對照溶液(75%乙醇)各適量,按“2.8.1”項下色譜條件進樣測定,記錄色譜圖。結果,刺桐堿峰和王不留行黃酮苷峰的分離度均大于1.5、理論板數均大于6 000,陰性對照不干擾測定,詳見圖6。
2.8.3 線性關系考察精密稱取刺桐堿對照品、王不留行黃酮苷對照品各適量,加甲醇制成每1 mL含刺桐堿321.832 μg、王不留行黃酮苷386.437 μg的混合對照品溶液。分別取該溶液1 mL,分置于1、2、5、10、25、50 mL量瓶中,加甲醇稀釋至刻度,搖勻,得系列線性工作溶液。分別精密吸取上述線性工作溶液1 μL,按“2.8.1”項下色譜條件進樣測定,記錄峰面積。以各待測成分質量濃度(x,μg/mL)為橫坐標、峰面積(y)為縱坐標進行線性回歸,得回歸方程,詳見表6。
2.8.4 定量限、檢測限考察 分別精密吸取“2.2”項下混合對照品溶液適量,加甲醇倍比稀釋后按“2.8.1”項下色譜條件進樣測定,分別以信噪比10 ∶ 1、3 ∶ 1計算定量限、檢測限,結果見表6。
2.8.5 精密度試驗 取“2.2”項下混合對照品溶液適量,按“2.8.1”項下色譜條件進樣測定6次,記錄峰面積。結果,刺桐堿和王不留行黃酮苷峰面積的RSD分別為0.16%、0.45%(n=6),表明儀器精密度良好。
2.8.6 重復性試驗 取王不留行樣品(編號:S3)適量,共6份,分別按“2.3”項下方法制備供試品溶液,按“2.8.1”項下色譜條件進樣測定,并按標準曲線法計算樣品中2種成分的含量。結果,刺桐堿、王不留行黃酮苷的平均含量分別為0.13%、0.52%,RSD分別為1.35%、1.78%(n=6),表明該方法重復性良好。
2.8.7 穩定性試驗 取“2.8.6”項下供試品溶液(編號:S3)適量,分別于室溫下放置0、2、4、6、12 h時按“2.8.1”項下色譜條件進樣測定,記錄峰面積。結果,刺桐堿、王不留行黃酮苷峰面積的RSD分別為0.53%、0.52%(n=5),表明供試品溶液于室溫下放置12 h內穩定性良好。
2.8.8 加樣回收率試驗 取王不留行生品(編號:S3)共6份,每份約0.5 g,精密稱定,分別加入一定量的刺桐堿、王不留行黃酮苷對照品,按“2.3”項下方法制備供試品溶液,再按“2.8.1”項下色譜條件進樣測定,記錄峰面積并計算加樣回收率。結果表明該方法準確度良好,詳見表7。
2.8.9 耐用性試驗 取王不留行生品(編號:S3)適量,按“2.3”項下方法制備供試品溶液,按“2.8.1”項下色譜條件以不同流速(0.30、0.35、0.40 mL/min)、不同柱溫(30、35、40 ℃)、不同色譜柱[Waters BEH C18(100 mm×2.1 mm,1.7 μm)、YMC C18(100 mm×2.1 mm,1.9 μm)、Agilent SB C18(100 mm×2.1 mm,1.8 μm)]進樣測定,記錄峰面積并按標準曲線法計算樣品中2種成分的含量。結果,刺桐堿、王不留行黃酮苷含量的RSD均小于3%(n=3),表明該方法能夠滿足試驗要求,耐用性良好。
2.8.10 樣品含量測定 取17批王不留行生品和17批炒王不留行各適量,分別按“2.3”項下方法制備供試品溶液,按“2.8.1”項下色譜條件進樣測定,記錄峰面積并按標準曲線法計算刺桐堿和王不留行黃酮苷的含量,結果見表8。由表8可知,王不留行生品中刺桐堿和王不留行黃酮苷的含量均比炒王不留行高,說明這2種成分在炮制后出現明顯下降,與周國洪等[14]的研究結果一致。
3 討論
本研究分別考察了不同色譜柱、流動相系統、檢測波長等色譜條件,經比較發現YMC Trait C18色譜柱(100 mm×2.1 mm,1.9 μm)、乙腈-水(梯度洗脫)和檢測波長為219 nm的分離效果較好、峰形更佳,故確定其為最終的色譜條件。
王不留行生品長于消癰腫;炒制后走散力較強,長于活血通經、下乳、通淋[16]。王不留行炮制前后功效的差異,說明炮制后王不留行所含化學成分發生了改變。中藥炮制后的變化主要體現在成分含量及成分種類的變化上,僅通過單一成分王不留行黃酮苷的含量作為指標性成分來控制王不留行炒制前后的質量有一定局限性。為此,本研究建立了鑒別、評價王不留行生品和炒王不留行的UPLC指紋圖譜,其相似度評價結果顯示,17批王不留行生品及相應的炮制品飲片的指紋圖譜的相似度均大于0.99,說明王不留行經炮制后其化學成分并未發生質的變化。通過聚類分析可以將王不留行生品和炒王不留行分為2類;在主成分分析中,可知色譜峰1(刺桐堿)和峰2(王不留行黃酮苷)是區分王不留行生品和炒王不留行的主成分,故對這2種成分進行含量測定,結果發現王不留行炮制后其化學成分發生了量的變化。從王不留行炮制前后的指紋圖譜并未發現有新成分產生,但是從峰面積結果發現,炮制后色譜峰1、2、4、5的峰面積均有一定幅度的減少,而色譜峰3的峰面積有所增加。由于色譜峰3、4、5均未能夠指認,色譜峰3與其余各峰之間是否存在轉化關系,則需要進一步對該色譜峰進行指認。此外,王不留行炮制前后化學成分的變化對藥理作用的影響尚需進一步開展藥效學實驗,對二者藥效作用的差異進行研究,以更好指導臨床合理用藥。
綜上所述,本研究建立了王不留行生品及其炮制品的UPLC指紋圖譜,該方法穩定、簡便、快速,結合相似度評價、聚類分析與主成分分析,可用于王不留行生品及其炮制品的質量評價。
參考文獻
[ 1 ] 國家藥典委員會.中華人民共和國藥典:一部[S]. 2015年版.北京:中國醫藥科技出版社,2015:54-55.
[ 2 ] 汪晶晶,任紅立,武洪志,等.中藥王不留行的化學成分及藥理作用研究進展[J].黑龍江畜牧獸醫,2017(4上):101-103.
[ 3 ] 魏薇.中藥王不留行的研究進展[J].中國醫藥指南,2014,12(16):87-88.
[ 4 ] 孟賀,陳玉平,秦文杰,等. HPLC 測定王不留行中王不留行黃酮苷的含量[J].中國中藥雜志,2010,35(16):2072-2074.
[ 5 ] 周國洪.王不留行化學成分及炮制對其影響研究[D].北京:中國中醫科學院中醫研究所,2016.
[ 6 ] 秦君,李慶章,高學軍.王不留行主要成分對小鼠乳腺上皮細胞增殖及β-酪蛋白表達的影響[J].中國農業科學,2008,41(8):2442-2447.
[ 7 ] 謝鳳珊,馮磊,馬麗萍,等.王不留行黃酮苷對過氧化氫和高糖誘導損傷的人臍靜脈內皮細胞的保護作用[J].天然產物研究與開發,2014,26(7):1009-1013.
[ 8 ] 王旭,候豹,蔡維維,等.王不留行黃酮苷的分離純化及對細胞增殖活性的影響[J].天然產物研究與開發,2017,29(2):316-321.
[ 9 ] 蔡維維,侯豹,陳旭紅,等.王不留行中刺桐堿的分離鑒定及抗炎活性研究[J].天然產物研究與開發,2018,30(4):616-620.
[10] 張亞麗,王東青.果實種子類藥物炮制淺見[J].遼寧中醫藥大學學報,2009,11(6):216.
[11] 郭建民.“逢子必炒”之探索[J].貴陽中醫學院學報,1994,16(3):59-60.
[12] 李翠芹,任鈞.王不留行生品與炮制品脂溶性成分的GC-MS分析[J].中成藥,2009,31(1):79-81.
[13] 周國洪,唐力英,寇真真,等.炮制對王不留行中王不留行環肽A,B,E含量的影響[J].中國實驗方劑學雜志,2016,22(4):29-31.
[14] 周國洪,唐力英,寇真真,等.炮制對王不留行中刺桐堿及黃酮苷類成分含量及溶出率的影響[J].中國實驗方劑學雜志,2016,22(22):18-21.
[15] 陰鉞玲,薛睿,孫兆林. UPLC法測定炮制前后王不留行中Vaccarin含量[J].亞太傳統醫藥,2015,11(20):20-21.
[16] 龔千鋒.中藥炮制學[M].北京:中國中醫藥出版社,2003:101.
(收稿日期:2020-05-14 修回日期:2020-07-02)
(編輯:胡曉霖)
猜你喜歡
四川蠶業(2021年2期)2021-03-09 03:15:32
四川蠶業(2021年3期)2021-02-12 02:38:46
中成藥(2018年11期)2018-11-24 02:57:00
中成藥(2017年8期)2017-11-22 03:19:40
中成藥(2017年10期)2017-11-16 00:50:13
中成藥(2017年4期)2017-05-17 06:09:50
哈爾濱醫藥(2016年1期)2017-01-15 13:43:16
天然產物研究與開發(2016年11期)2016-06-15 20:29:17
湖南師范大學自然科學學報(2015年1期)2015-02-27 14:50:04
安徽醫藥(2014年12期)2014-03-20 13:15:15
