槲皮素磁性分子印跡聚合物的制備
2020-10-26 08:51:28朱安宏涂清波
化學(xué)與生物工程 2020年10期
關(guān)鍵詞:功能
朱安宏,秦 政,涂清波
(南京中醫(yī)藥大學(xué)翰林學(xué)院藥學(xué)院,江蘇 泰州 225300)
槲皮素(quercetin,Qu)是一種含有多種生物活性的天然黃酮醇類化合物,在抗氧化、抑制腫瘤、抗菌、抗炎、防治糖尿病并發(fā)癥等方面具有重要作用。天然產(chǎn)物中的槲皮素濃度很低,對其進行分離與提純相對困難。近年來,分子印跡技術(shù)被廣泛應(yīng)用于天然產(chǎn)物活性成分的提取[1-3]。分子印跡聚合物(MIPs)由于對模板分子具有選擇性識別的特性,受到研究者越來越多的關(guān)注。目前,采用分子印跡聚合物萃取槲皮素的研究較多[4],但是在聚合物的吸附過程中,將目標物從復(fù)雜體系中分離需要反復(fù)多次抽濾和離心,分離效率較低。磁性納米材料[5-8],特別是穩(wěn)定、廉價、易制備的Fe3O4,由于具有良好的磁學(xué)性能及穩(wěn)定的物理和化學(xué)性質(zhì)而被廣泛應(yīng)用于各個分離領(lǐng)域,可以實現(xiàn)從復(fù)雜的基體中迅速分離目標物。因此,磁性納米材料結(jié)合分子印跡技術(shù)的研究也越來越多[9-11]。
作者以Fe3O4磁性納米顆粒為載體、槲皮素為模板分子、丙烯酰胺(AM)為功能單體、無水乙醇為致孔劑,通過沉淀聚合法制備槲皮素磁性分子印跡聚合物(Qu/MMIPs),采用掃描電鏡和透射電鏡對其形貌進行表征,并考察其吸附性能。
1 實驗
1.1 試劑與儀器
槲皮素二水合物(98%)、丙烯酰胺(AM,98%)、2,2-偶氮二異丁腈(AIBN,98%)、二甲基丙烯酸乙二醇酯(EGDMA,98%),上海阿拉丁試劑有限公司;乙二醇(EG)、聚乙二醇4000(PEG4000)、六水三氯化鐵(FeCl3·6H2O),分析純,國藥集團化學(xué)試劑有限公司;超純水;其它試劑均為分析純。
KQ500B型超聲波清洗儀,昆山超聲儀器有限公司;ME104E型電子分析天平,梅特勒-托利多儀器(上海)有限公司;DHG-914385-Ⅲ型電熱恒溫鼓風干燥箱,上海新苗醫(yī)療器械制造有限公司;DF-202B型集熱式恒溫加熱磁力攪拌器,上海凌科實業(yè)發(fā)展有限公司;TGL-16C型高速臺式離心機,上海安亭科學(xué)儀器廠;UC-1800PC型紫外可見分光光度計,上海元析儀器有限公司;聚四氟乙烯不銹鋼高壓反應(yīng)釜(100 mL),上海秋佐科學(xué)儀器有限公司;Dragon移液槍;索式提取器等。
1.2 Fe3O4磁性納米顆粒的制備
采用水熱法[12]制備Fe3O4磁性納米顆粒。取20 mL EG溶液置于聚四氟乙烯不銹鋼高壓反應(yīng)釜中,加入1.02 g FeCl3·6H2O、2.82 g CH3COONa、0.086 g PEG4000,溶解,攪拌均勻后,擰緊反應(yīng)釜,密閉加熱至200 ℃,反應(yīng)12 h;取出后冷卻至室溫,依次用蒸餾水、無水乙醇超聲洗滌黑色產(chǎn)物5 min,用磁石吸住黑色產(chǎn)物,將洗液除去;反復(fù)洗滌數(shù)次后,60 ℃下真空干燥24 h,即得Fe3O4磁性納米顆粒。
1.3 槲皮素與功能單體的相互作用
1.3.1 溶液的配制
精密稱取3.38 mg(0.01 mmol)槲皮素,置于100 mL容量瓶中,加入適量乙醇,超聲使其充分溶解,冷卻至室溫后用乙醇定容至刻度,得濃度為0.1 mmol·L-1的槲皮素乙醇溶液,備用。精密稱取0.01 mmol功能單體AM、甲基丙烯酸(MAA)和乙烯基吡啶(4-VP),分別置于100 mL容量瓶中,加入適量乙醇,超聲使其充分溶解,冷卻至室溫后用乙醇定容至刻度,得濃度均為0.1 mmol·L-1的AM、MAA和4-VP溶液,備用。
1.3.2 槲皮素與功能單體配比的選擇
分別按槲皮素與功能單體AM配比為1∶0、1∶2、1∶4、1∶6、1∶8配制混合標準溶液,靜置3 h使其充分作用,用紫外分光光度計測定300~500 nm范圍內(nèi)的紫外光譜,確定適宜的配比。
1.3.3 功能單體的選擇
以槲皮素為模板分子、無水乙醇為致孔劑,在不改變交聯(lián)劑和引發(fā)劑用量的條件下,分別以AM、MAA和4-VP為功能單體,按模板分子與功能單體配比為1∶2、1∶4、1∶6制備分子印跡聚合物MIP1~MIP9,測定MIP1~MIP9對槲皮素的吸附量,確定適宜的功能單體。
1.4 Qu/MMIPs、MNIPs的制備
采用沉淀聚合法制備Qu/MMIPs。準確稱取0.033 8 g(0.1 mmol)槲皮素模板分子、0.021 3 g(0.3 mmol)功能單體、500 mg Fe3O4磁性納米顆粒置于裝有50 mL無水乙醇的圓底燒瓶中,超聲5 min混合均勻,室溫下反應(yīng)3 h;再加入20 mmol交聯(lián)劑EGDMA、30 mg引發(fā)劑AIBN,通入氮氣20 min后密封,80 ℃反應(yīng)24 h;以甲醇-乙酸(9∶1,體積比)混合溶液為溶劑,采用索氏提取法洗脫產(chǎn)物,直至洗脫液中無槲皮素的紫外吸收,即分子印跡聚合物中模板分子已去除,60 ℃干燥過夜,即得Qu/MMIPs。
不加槲皮素模板分子,同法制備磁性非印跡聚合物MNIPs。
1.5 Qu/MMIPs的表征
采用掃描電鏡和透射電鏡對Fe3O4磁性納米顆粒、Qu/MMIPs、MNIPs的形貌進行表征;通過分析磁滯回線,對Fe3O4磁性納米顆粒、Qu/MMIPs的磁學(xué)性能進行表征。
1.6 Qu/MMIPs的吸附性能評價
1.6.1 吸附動力學(xué)實驗
吸附動力學(xué)實驗?zāi)芊从尘酆衔锏奈搅颗c吸附時間之間的關(guān)系[7]。分別準確稱取500 mg Qu/MMIPs和MNIPs置于250 mL錐形瓶中,加入100 mL 0.01 mg·mL-1的槲皮素乙醇溶液,放入水浴振蕩器中,常溫下分別振蕩20 min、40 min、60 min、80 min、120 min、180 min、240 min、300 min、360 min,取混合液1 mL,微孔過濾后稀釋至5 mL,用紫外分光光度計測定其在254 nm處的吸光度,根據(jù)朗伯比爾定律確定槲皮素的平衡濃度。按下式計算聚合物對槲皮素的吸附量(Q,mg·g-1),并繪制吸附量與吸附時間(t)的動力學(xué)吸附曲線。
式中:c0為槲皮素的初始濃度,mg·mL-1;ct為吸附平衡時槲皮素的濃度,mg·mL-1;V為槲皮素溶
液的體積,mL;m為Qu/MMIPs或MNIPs的質(zhì)量,g。
1.6.2 等溫吸附曲線的繪制
分別配制濃度(mg·mL-1)為0.01、0.02、0.04、0.06、0.08、0.10、0.12、0.14的槲皮素乙醇溶液,準確移取5 mL置于25 mL容量瓶中,加入50 mg Qu/MMIPs或MNIPs,用無水乙醇定容至刻度,常溫下振蕩3 h,離心,移取1 mL上清液稀釋5倍后用紫外分光光度計測定其在254 nm處的吸光度,按1.6.1方法計算Qu/MMIPs和MNIPs對槲皮素的吸附量,繪制等溫吸附曲線,并依據(jù)吸附量評價Qu/MMIPs和MNIPs的吸附性能。
2 結(jié)果與討論
2.1 槲皮素和功能單體配比的選擇
槲皮素與AM相互作用的紫外光譜如圖1所示。
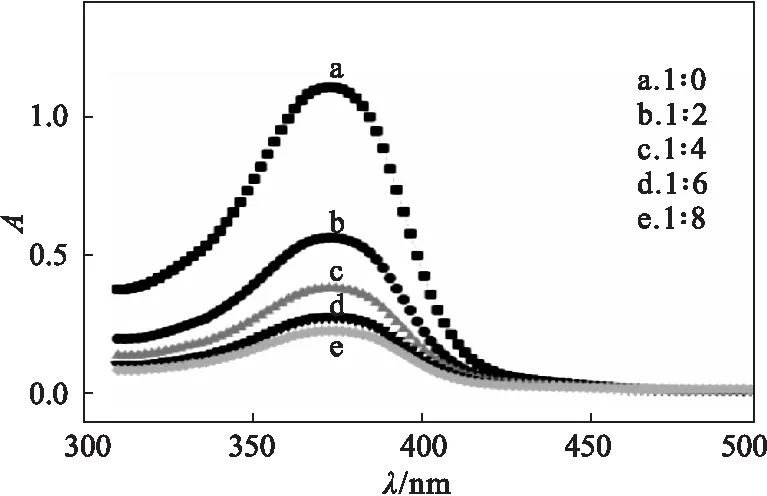
圖1 槲皮素與AM相互作用的紫外光譜Fig.1 UV spectra for interaction between quercetin and AM
從圖1可以看出,隨著功能單體AM占比的增加,槲皮素和功能單體AM混合標準溶液的吸光度呈下降趨勢,表明槲皮素與功能單體AM間的相互作用影響了槲皮素對紫外光的吸收,而且相互作用越大,吸光度變化量(ΔA)越大。當槲皮素與AM配比為1∶6時,相互作用趨于穩(wěn)定,繼續(xù)增加AM占比,對吸光度的影響較小。因此,選擇槲皮素與AM的最佳配比為1∶6。
2.2 功能單體的選擇
分子印跡聚合物MIP1~MIP9對槲皮素的吸附量見表1。
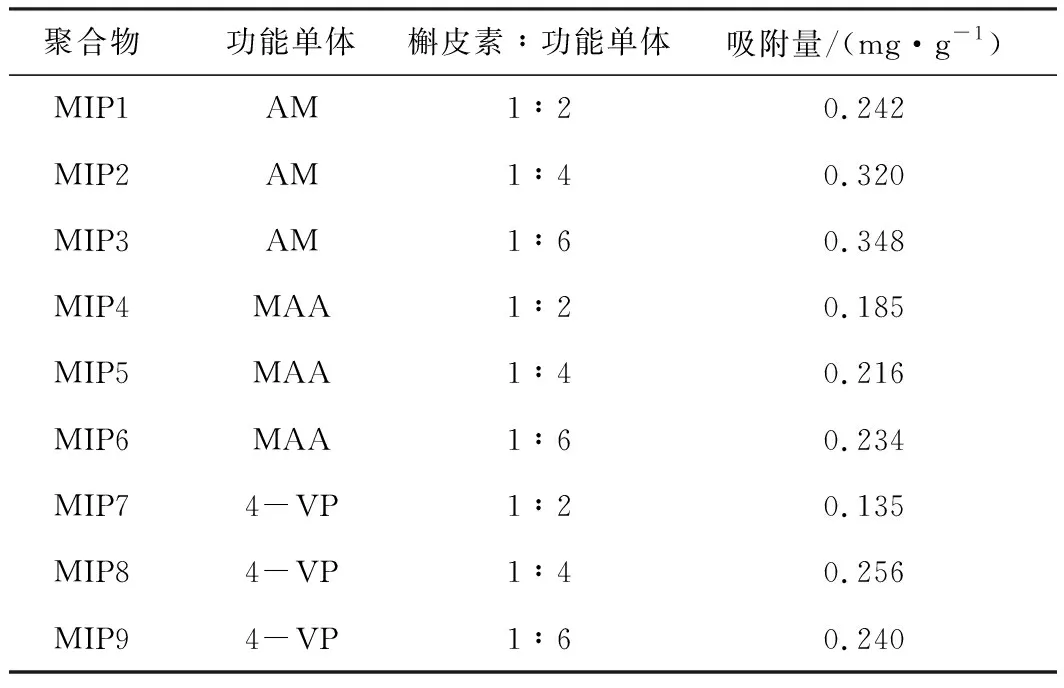
表1 分子印跡聚合物對槲皮素的吸附量
從表1可知,以MAA和4-VP為功能單體制備的分子印跡聚合物MIP4~MIP9對槲皮素的吸附量比以AM為功能單體制備的MIP1~MIP3小。主要是由于,槲皮素與AM之間的相互作用更強,兩者之間形成更多的氫鍵。當槲皮素與AM配比為1∶6時,吸附量最大。這與配比選擇實驗結(jié)果相符。
2.3 形貌表征(圖2)

圖2 Qu/MMIPs(a)、MNIPs(b)、Fe3O4磁性納米顆粒(c)的SEM照片及Qu/MMIPs的TEM照片(d)Fig.2 SEM images of Qu/MMIPs(a),MNIPs(b),Fe3O4 magnetic nanoparticles(c),and TEM image of Qu/MMIPs(d)
從圖2可以看出,Qu/MMIPs與MNIPs都呈球狀結(jié)構(gòu)團簇在一起,粒徑基本一致;Fe3O4磁性納米顆粒粒徑均一,約為600 nm。與Fe3O4磁性納米顆粒粒徑相比,Qu/MMIPs的粒徑變大(圖2d),表明Fe3O4磁性納米顆粒的表面包裹著分子印跡聚合物,成功制備了以Fe3O4磁性納米顆粒為核心的磁性分子印跡聚合物。
2.4 磁學(xué)性能(圖3)
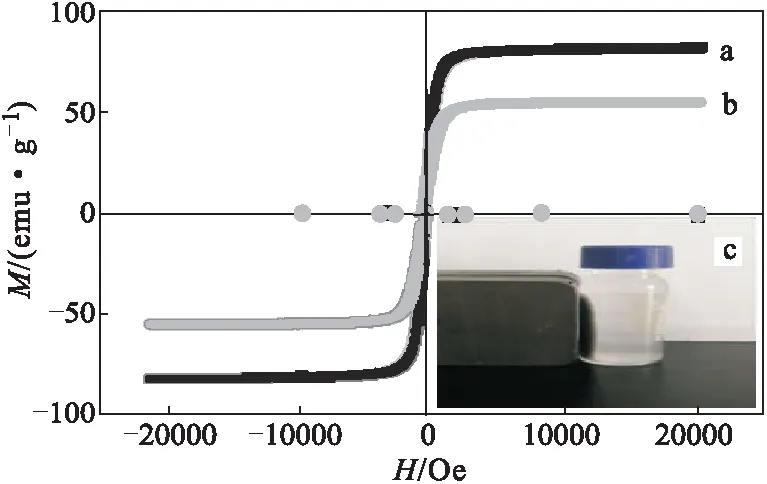
圖3 Fe3O4磁性納米顆粒(a)、Qu/MMIPs(b)的磁滯回線及Qu/MMIPs在外磁場下的磁分離現(xiàn)象照片(c)Fig.3 Hysteresis loops of Fe3O4 magnetic nanoparticles(a),Qu/MMIPs(b) and photo for Qu/MMIPs magnetic separation phenomenon in external magnetic field(c)
從圖3可以看出,F(xiàn)e3O4磁性納米顆粒和Qu/MMIPs的磁滯回線都是可逆的,不存在磁滯現(xiàn)象,其表征物理量矯頑力以及剩磁的數(shù)值大小幾乎為零,在外磁場的作用下順磁磁化率高于一般材料,展現(xiàn)出良好的超順磁性。Fe3O4磁性納米顆粒、Qu/MMIPs的飽和磁化強度分別為80.8 emu·g-1、54.3 emu·g-1,Qu/MMIPs的飽和磁化強度低是因為,在聚合過程中Fe3O4磁性納米顆粒的表面包裹了一層分子印跡聚合物,對磁響應(yīng)產(chǎn)生了一些影響,但并不影響Qu/MMIPs的磁學(xué)性能。當外部環(huán)境有磁場存在時,Qu/MMIPs能夠迅速聚集到靠近磁鐵的一邊,溶液變得澄清透明(圖3c),從而可以迅速地將目標物從復(fù)雜基體中分離。Qu/MMIPs具有良好的磁效應(yīng),在外加磁場作用下分離效果明顯,同時該方法制備的磁性分子印跡聚合物也適用于其它化學(xué)成分的富集分離。
2.5 動力學(xué)吸附曲線(圖4)
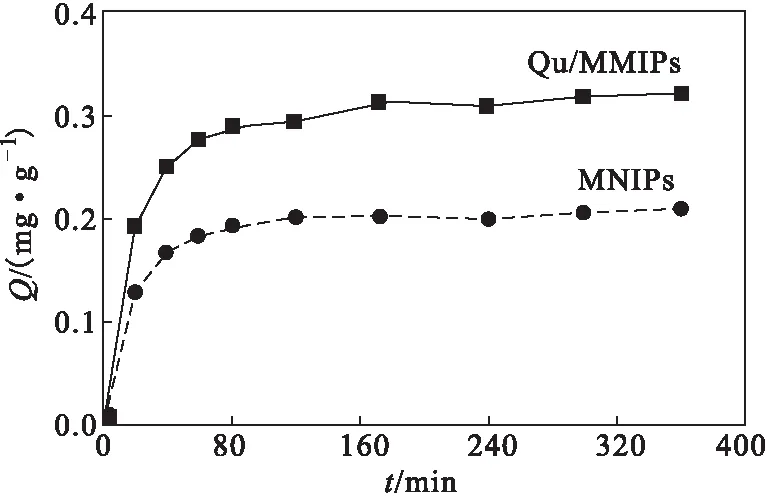
圖4 動力學(xué)吸附曲線 Fig.4 Dynamic adsorption curves
從圖4可以看出,Qu/MMIPs和MNIPs對槲皮素的吸附量隨吸附時間的延長而增大,特別是在初期,吸附量急劇增大,表明初期時的吸附速率很快,主要是由于Qu/MMIPs和MNIPs的非特異性吸附作用所致。Qu/MMIPs的整體吸附量大于MNIPs的,這是由于,MNIPs無印跡過程,聚合物表面沒有特異性識別位點,對槲皮素的吸附屬于物理吸附,即非特異性吸附;而Qu/MMIPs的表面存在大量的印跡位點,能與槲皮素在空間結(jié)構(gòu)和官能團作用相匹配,對槲皮素特異性識別和吸附。40 min后,由于聚合物表面大量的印跡位點被槲皮素分子占據(jù),槲皮素分子很難進入聚合物內(nèi)部,傳質(zhì)阻力增大,從而使吸附速率變慢;60 min后,吸附量基本無變化,吸附達到平衡。
2.6 等溫吸附曲線(圖5)
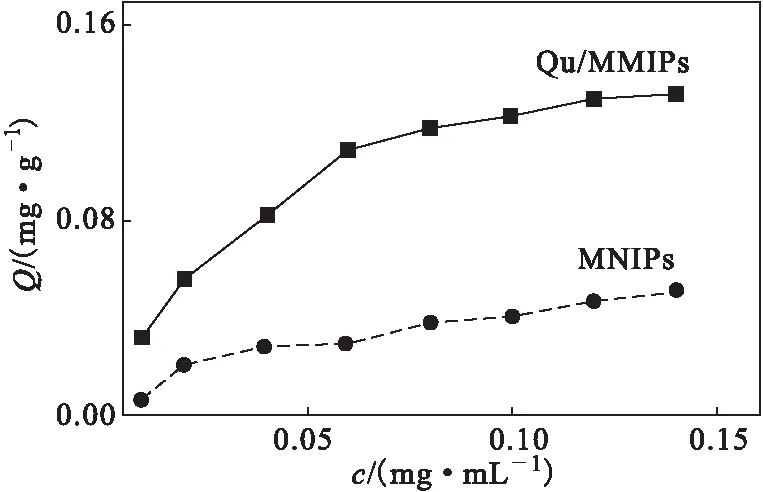
圖5 等溫吸附曲線Fig.5 Isothermal adsorption curves
從圖5可以看出,Qu/MMIPs、MNIPs對槲皮素的吸附量均隨濃度的增加而增大,且Qu/MMIPs的吸附量大于MNIPs的;在濃度為0.04 mg·mL-1時,MNIPs對槲皮素的吸附趨于平衡,非特異性吸附達到飽和,吸附量基本不變。除了非特異性吸附外,Qu/MMIPs的表面還有大量對槲皮素特異性識別的印跡位點,且其內(nèi)部也有很多印跡位點,又因為Qu/MMIPs內(nèi)部的傳質(zhì)阻力較大,因此,Qu/MMIPs對槲皮素的吸附量雖然逐漸增大,但吸附速率逐漸減慢直到吸附平衡。
3 結(jié)論
以Fe3O4磁性納米顆粒為載體、槲皮素為模板分子、AM為功能單體、無水乙醇為致孔劑,通過沉淀聚合法制備了Qu/MMIPs。Qu/MMIPs具有較好的磁學(xué)性能且對槲皮素模板分子具有特異選擇性,可以從復(fù)雜的天然產(chǎn)物體系中快速有效地分離槲皮素。為天然產(chǎn)物中其它化學(xué)成分的分離純化提供了一種新方法。
猜你喜歡
鐘表(2023年5期)2023-10-27 04:20:44
中華詩詞(2022年6期)2022-12-31 06:41:24
當代陜西(2021年21期)2022-01-19 02:00:26
中學(xué)生數(shù)理化(高中版.高考數(shù)學(xué))(2020年1期)2020-02-20 13:23:44
經(jīng)濟技術(shù)協(xié)作信息(2018年11期)2019-01-14 03:07:20
中國科技論壇(2017年7期)2017-07-25 08:49:53
制造技術(shù)與機床(2017年3期)2017-06-23 08:11:33
媽媽寶寶(2017年2期)2017-02-21 01:21:24
國際漢語學(xué)報(2016年1期)2017-01-20 08:21:20
中國中醫(yī)藥現(xiàn)代遠程教育(2014年22期)2014-03-01 04:32:55
