竹材在LiCl/DMSO溶劑體系中的溶解及再生性能
2020-10-17 03:04:00吳文娟鄒春陽黃麗菁金永燦
林業科學 2020年9期
吳文娟 鄒春陽 黃麗菁 金永燦
(南京林業大學 江蘇省林業資源高效加工利用協同創新中心 南京 210037)
木材和非木材植物纖維原料的主要化學成分為纖維素、半纖維素和木質素,占原料總質量的80%~95%,其中,木質素為高度交聯的芳香族高聚物,具有膠黏作用,將纖維素和半纖維素相互連接在一起,并給予加固硬質化,以避免微生物侵蝕到細胞壁(Kammetal., 2006)。在木質素大分子結構中,作為木質素的基本單元,苯丙烷以醚鍵或碳-碳鍵的連接方式組成交叉網絡的立體結構,作為大分子,其結構中也有以化學鍵的連接方式與碳水化合物結合在一起,形成具有立體網狀結構的大分子,并以木質素-碳水化合物的復合物(LCC)形式存在于植物細胞壁中。木質纖維原料結構復雜致密,在生物或化學降解上具有明顯抑制作用,且錯綜交織結構的存在也不利于分出全部原本木質素,從而無法獲取到木質素的相關原始結構信息(Adler, 1977; Leeetal., 1981; Changetal., 1975)。因此,有效抑制或消除這種阻礙,對發展、實施木質纖維原料的高度生物煉制具有重大意義。
目前,對原本木質素的了解主要基于一些傳統、經典的方法,如球磨木質素(MWL)、纖維素酶解木質素(CEL),這些方法均需要一定程度的球磨處理,但球磨程度不同會導致木質素結構不可避免的破壞(Ikedaetal., 2002; Fujimotoetal., 2005; Huetal., 2006; Guerraetal., 2006; Holtmanetal., 2004),將模糊對原本木質素最初結構的認識。盡管研究人員在這一方面做了大量工作,但通過分離得到的各組分會發生一定程度的降解或衍生化反應,使得供分析的各組分結構特征不具備完全公認的代表性。對于木質素所在載體——木質纖維原料,由于其本身組成結構復雜,要了解彼此之間的組成連接,當前除了合適的分離手段外,還可借助木質纖維原料的全溶體系,利用高分辨率二維核磁共振技術對細胞壁組分進行結構鑒定,分析速度快,且可獲得更加全面的各組分結構信息。在全溶體系下,NMR的光譜分析可給出完整木質素的結構信息,這是分離出來的木質素無法提供的; 但并不是所有全溶體系都具有這樣的特點,有些體系在溶解過程中可能會有衍生物生成(Kuboetal., 2008; Heinzeetal., 2000),如有機溶劑復合體系二甲基亞砜/四丁基銨(DMSO/TBAF),細胞壁組分在全溶體系中發生乙酰化反應后,會伴隨其他組分的衍生; 有些全溶體系需要在苛刻條件下進行(Kilpel?inenetal., 2007),如咪唑類溶劑,將木片完全溶解,需要在150 ℃下持續溶解24 h; 也可能與原料的球磨程度有關(Luetal., 2003)。這些都可能改變原料中各種初始化學組成的變化,從而會影響初始結構的信息收集。
LiCl/DMSO(氯化鋰/二甲基亞砜)溶劑體系一直作為高聚物的反應溶劑(Petru?etal., 1995)。Wang等(2009)將該溶劑體系用于木材溶解,成功得到了不同于上述的木質纖維素全溶體系。LiCl/DMSO溶劑體系可有效改善球磨時間對木質素結構的影響,與其他木質纖維原料溶劑體系相比,該體系的最大優點是制備球磨樣品時,球磨時間可縮短至2 h,且全溶時樣品質量分數高達10%。LiCl/DMSO溶劑體系所得木質纖維素全溶組分能夠直接用紫外、核磁共振等進行分析,相繼用于稻草、麥草等非木材木質纖維原料的溶解以及木質素的分離和表征(Wuetal., 2014a; Guetal., 2015; Caoetal., 2016; Jiangetal., 2017)。本研究以竹材為原料,探索竹材在LiCl/DMSO溶劑體系中的溶解行為及再生特點,以期為全面解析竹材原本木質素結構信息提供一個可行的木質素分離溶劑體系。
1 材料與方法
1.1 原料與試劑 試驗竹材——毛竹(Phyllostachysedulis)產自浙江安吉,人工劈成長3~4 cm,寬、厚 2~4 mm 的小竹條。風干后用Wiley微型粉碎機粉碎,取40~80目組分,用苯醇(2∶1,V/V)抽提8 h,經真空干燥后得脫脂竹粉(B),貯存于帶磨砂玻璃塞的廣口瓶中,供分析使用。本研究所用試劑均為分析純。
1.2 球磨 球磨采用德國Fritsch微型行星式高能球磨機。稱取3 g真空干燥后的脫脂竹粉裝入容積為45 mL的氧化鋯制罐子中,內裝18只內徑1 cm的氧化鋯球,在600 r·min-1下根據需要進行不同時間球磨。球磨在冷庫(-20 ℃)進行,每運行15 min休停5 min,以避免過熱。球磨竹粉經干燥后備用。
1.3 LiCl/DMSO溶劑體系的溶解 配制不同質量分數(2%、4%、6%、8%等)的LiCl/DMSO溶液。稱取不同球磨時間的竹粉,在LiCl/DMSO溶劑體系中(室溫)進行溶解處理。
1.4 LiCl/DMSO溶劑體系的再生 LiCl/DMSO溶劑體系處理脫脂原料的流程如圖1所示。球磨竹粉在6% LiCl/DMSO溶劑體系中進行溶解處理,完全溶解后,整個混合體系全部轉移到孔徑50 ?、直徑28 mm、透過分子量14 000的透析袋,并將透析袋浸沒于水中,用去離子水透析,用AgNO3溶液檢測有無Cl-來判斷是否完全置換出LiCl/DMSO溶劑體系。袋內物料經冷凍干燥后得到球磨的再生原料(RB)。
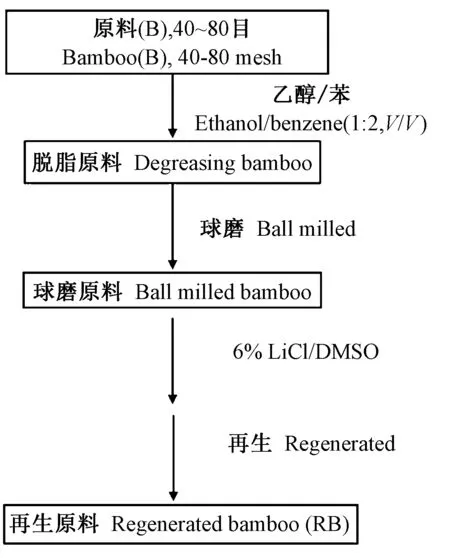
圖1 經LiCl/DMSO溶劑體系處理后的再生流程Fig.1 Procedure of regeneration by LiCl/DMSO
1.5 分析方法 1) 化學成分測定 木質素含量參照文獻(Denceetal., 1992)方法測定。稱取樣品0.3 g(精確到0.000 1 g),加入3 mL質量分數為72%的H2SO4,18~20 ℃下水解 3 h,期間定時攪拌,然后轉入100 mL玻璃瓶中,加水將H2SO4稀釋至質量分數4%,密封后置于滅菌鍋(DSX-280B)內,121 ℃下水解 1.5 h。采用恒重的G3砂芯漏斗分離殘渣和濾液,收集濾液測定酸溶木質素和高聚糖含量。殘渣用熱蒸餾水洗滌,濾液用質量分數10%的BaCl2檢測無白色沉淀,在恒溫干燥箱中105 ℃干燥至恒重,稱量后轉入馬弗爐中 575 ℃灼燒至恒重,測定木質素中灰分含量。計算 Klason 木質素含量時扣除灰分含量。以質量分數4%的H2SO4為參比,用日本島津公司UV-240紫外-可見分光光度計測定濾液在波長 205 nm處的吸光度,計算酸溶木質素含量。還原糖含量測定參照文獻(Borchardtetal., 1970)。取濾液5 mL,加入內標1 mL肌醇溶液(1 mg·mL-1),用飽和Ba(OH)2溶液調節pH至5.5,離心分離除去沉淀,加入20 mg NaBH4于清液中,靜置24 h。加入1 mL醋酸,旋轉蒸發至漿狀,再加入甲醇繼續蒸發至干,每次1 mL,連續3次。蒸干體系于105 ℃烘干15 min,以確保體系完全無水。加入1 mL乙酸酐后密封,120 ℃反應3 h。用日本島津公司GC-14b氣相色譜儀分析還原糖。氣相色譜條件如下: 毛細管柱TC-17 (0.25 mm×30 m); FID檢測器; 柱溫程序,220 ℃保留20 min,進樣溫度220 ℃,檢測溫度230 ℃。GC分析得到的單糖均轉換為高聚糖。同時進行2次測定,取平均值。
2) 硝基苯氧化 木質素結構單元變化通過堿性硝基苯氧化分析。硝基苯氧化操作參照文獻(Chen, 1992)。取樣品40 mg于10 mL不銹鋼制罐子內,依次加入硝基苯0.25 mL、2 mol ·L-1NaOH 4 mL,將上述混合體系密封置于170 ℃油浴中反應2 h。冷卻至室溫,加入1 mL 0.1 mol ·L-1NaOH配制的內標3-乙氧基-4-羥基苯甲醛(0.2~0.4 g·L-1)。用二氯甲烷萃取3次,每次15 mL,棄油相取水相,用4 mol ·L-1HCl調節pH至1,再用二氯甲烷萃取2次,每次20 mL,乙醚1次,15 mL。收集有機相共55 mL,用20 mL去離子水洗滌有機相,經無水Na2SO4干燥,過濾,濾液蒸發至干,加入100 μL N, O-雙(三甲基硅)乙酰胺衍生化試劑(BSA)100 ℃反應10 min,產物用日本島津公司GC-17A氣相色譜儀進行分析。氣相色譜條件如下: 毛細管柱NB-1 (0.25 mm×30 m); FID檢測器; 柱溫程序,150 ℃保留15 min,以3 ℃·min-1升溫到180 ℃,繼而以10 ℃·min-1升溫到280 ℃,保留5 min; 進樣溫度250 ℃,檢測溫度280 ℃。同時進行2次測定,取平均值。
3) X射線衍射 纖維結晶指數分析在日本理學Ultima IV組合型多功能水平X射線衍射儀上進行,采用粉末法。測試條件: X光管為CuKα靶(λ=0.154 18 nm),用石墨單色器消除CuKα輻射,管電壓40 kV,管電流40 mA,采用θ/2θ聯動掃描,索拉狹縫0.04 rad,接收狹縫0.15 mm,發射和防散射狹縫1°,掃描范圍2θ=5°~40°,掃描步長0.02°,掃描速率5(°)·min-1,記錄“衍射強度-2θ”曲線。
結晶指數根據Segal公式(Segaletal., 1959)計算,如下:
式中: CrI為結晶指數;Iamorph為 2θ為18°時無定形區衍射峰強度;I002為主結晶峰 002 的最大衍射強度。
2 結果與討論
2.1 竹材在LiCl/DMSO溶劑體系中的溶解性能 1) 不同球磨時間對竹材溶解性能的影響 圖2為不同球磨時間竹粉(質量分數7.5%)室溫下攪拌6 h后在6% LiCl/DMSO溶劑體系中的溶解性能。可以看出,球磨時間不同,竹材在LiCl/DMSO溶劑體系中的溶解性能亦有所不同。未球磨的40~80目竹粉,在LiCl/DMSO溶劑體系中呈現混濁且有分層狀態,球磨0.5 h、1 h的竹粉不能完全溶解在6% LiCl/DMSO溶劑體系中,而球磨1 h的稻麥草能在該溶劑體系中完全溶解(Wuetal., 2014b; Guetal., 2015); 進一步球磨發現,球磨2 h的竹粉能在該溶劑體系中徹底溶解,可得到澄清透亮的溶液。相較于稻草原料,竹材材質更為緊致,接近于木材。LiCl/DMSO作為再生纖維的溶劑體系,其中LiCl在溶解再生纖維素時具有破壞纖維素氫鍵并阻止重構氫鍵的作用,以此促進再生纖維素進一步溶解(Petru?etal., 1995)。該溶劑體系雖然不適用于一般纖維素溶解,但在原料選擇上有進一步拓寬,即可作為球磨木粉的新型溶劑。與木材相比,禾草類纖維雖然有著化學成分、化學結構上不一樣的特點,但基于一定條件,非木材在該溶劑體系中仍具有類似的溶解性能。

圖2 不同球磨時間的竹材溶解性能(6% LiCl/DMSO)Fig.2 Effect of milling time in 6% LiCl/DMSO solvent system on dissolution for bamboo
2) LiCl/DMSO溶劑體系質量分數對竹材溶解性能的影響 圖3為球磨2 h的竹粉在不同質量分數LiCl/DMSO溶劑體系中的溶解性能。可以看出,隨著LiCl/DMSO溶劑體系質量分數增加,球磨竹粉在其中的溶解度亦隨之增加。球磨2 h的竹粉能夠完全溶解于6% LiCl/DMSO溶劑體系中,溶解后體系透明澄清,可溶解竹粉的上限量為7.5%(在LiCl/DMSO體系中的質量分數)。隨著LiCl/DMSO溶劑體系質量分數增加,球磨2 h的竹粉溶解度增大,在8%LiCl/DMSO溶劑體系中可溶解竹粉的質量分數達10%。與木材相比,LiCl/DMSO溶劑體系更易于溶解非木質纖維原料,可降低球磨時間及所需的溶解時間。

圖3 不同質量分數LiCl/DMSO溶劑體系的竹材溶解性能Fig.3 Effect of LiCl content in LiCl/DMSO solvent system on dissolution for bamboo
2.2 竹材在LiCl/DMSO溶劑體系中的再生性能 1) 再生竹材的化學成分 竹材在LiCl/DMSO溶劑體系中具有與木材類似的溶解性能,得到再生竹材的主要化學成分見表1。可以看出,再生竹材中Klason木質素有少許下降,這是因為經LiCl/DMSO溶劑體系處理后的竹粉在水中再生時采用的是孔徑50 ?、透過分子量14 000的透析袋,一些小分子量、小孔徑的成分會在再生過程中透過透析袋,進入到水中,最終被移除。相較于原料中的Klason木質素,球磨2 h的竹粉中Klason木質素下降3.2%,而酸溶木質素在LiCl/DMSO溶劑體系溶解再生過程中沒有變化。球磨2 h的竹粉中木質素得率比脫脂竹粉低10%,高聚糖得率為85%,球磨和LiCl/DMSO溶劑體系溶解再生對原料中高聚糖含量具有一定影響。經LiCl/DMSO溶劑體系處理,再生后竹材可更多地保留化學成分,其中高聚糖溶出量比木質素多,相較于在同樣溶劑體系中的球磨稻草粉(Wuetal., 2014b)、木粉(Wangetal., 2009),竹材在溶解性能上更接近于木材。
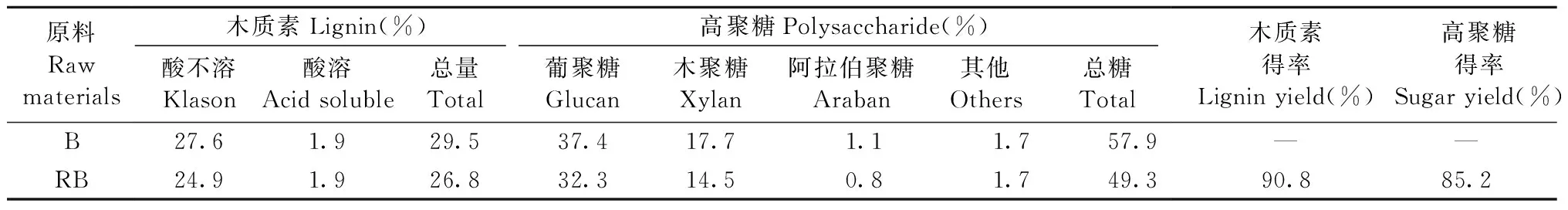
表1 再生竹材的主要化學成分(基于原料)Tab.1 Chemical compositions of bamboo and regenerated bamboo (based on raw materials)
2) 再生竹材木質素結構單元的變化 木質素結構中存在愈瘡木基丙烷、紫丁香基丙烷和對-羥基苯基丙烷3種類型的結構單元。針葉材木質素主要以愈創木基丙烷為主,闊葉材木質素除愈創木基丙烷外,還有較多的紫丁香基丙烷,禾草類木質素則是愈創木基丙烷、紫丁香基丙烷和對-羥基苯丙烷3種結構單元的混合物。木質素堿性硝基苯氧化產物可揭示木質素苯環單元構成,表2給出了球磨竹粉經LiCl/DMSO溶劑體系處理再生后硝基苯氧化產物得率及木質素結構單元變化。可以看出,雖然同屬于禾草類,但竹材在木質素化學結構上卻有著與稻草不一樣的特點,木質素中紫丁香基單元含量占3種結構單元之和的一半。與未經任何處理的原料竹材相比,球磨2 h的竹粉化學結構單元幾乎沒有變化,再生原料的硝基苯氧化產物得率及各結構單元含量經過球磨、LiCl/DMSO溶劑體系處理后也幾乎沒有變化。脫脂竹粉的硝基苯氧化產物包括對羥基苯甲醛(H)、香草醛(V)和紫丁香醛(S),總得率為2.5 mmol·g-1,其中S單元占50%,與文獻中竹材木質素結構單元含量及分布一致(黃曹興等, 2018); 竹粉經球磨、再生處理后,硝基苯氧化產物得率為2.4 mmol·g-1,S單元仍為50%,均高于稻草的得率(Wuetal., 2014b),即木質素的縮合程度不及稻草。由此可知,球磨及LiCl/DMSO溶劑體系處理對原料中木質素結構均沒有影響。

表2 竹材硝基苯氧化產物的得率及S/VTab.2 Nitrobenzene oxidation products yields and S/V molar ratio of bamboo samples

圖4 竹材原料(B)和再生原料(RB)的X射線衍射分析Fig.4 X-ray pattern of original bamboo and regenerated bamboo with ball milling
3) 再生處理對竹材纖維素結晶度的影響 圖4為竹材原料及經球磨、LiCl/DMSO溶劑體系處理后再生原料的X射線衍射曲線,表3為竹材原料和再生原料纖維素的結晶度。可以看出,球磨2 h并經LiCl/DMSO溶劑體系處理后再生原料的衍射曲線明顯不同于竹材原料,結合表3,原料再生后的結晶度由原來的58.7%降低至26.5%,說明LiCl/DMSO溶劑體系處理球磨原料會影響原料中纖維素的聚集態結構。纖維素本身結構復雜,內部存在大量的晶區結構、非晶區結構和氫鍵,溶劑對纖維素的可及度低,一般溶劑難以溶解,球磨后經LiCl/DMSO溶劑體系處理,會使原料中原有的結晶區域有一定程度破壞,從而有利于球磨竹粉在LiCl/DMSO溶劑體系中進一步溶出,同時LiCl/DMSO溶劑體系作為預處理所帶來的結晶區改變,有望達到提高酶解效率的作用。

表3 竹材原料和再生原料纖維素的結晶度Tab.3 Crystallinity of bamboo and regenerated bamboo with ball milling
3 結論
球磨2 h的竹粉能以質量分數7.5%完全溶解在6%LiCl/DMSO溶劑體系中,溶于8%LiCl/DMSO溶劑體系中的竹粉質量分數達10%; 再生后,球磨竹粉的化學成分再生能力強,木質素得率約90%,高聚糖在6%LiCl/DMSO溶劑體系的再生能力稍低于木質素。竹材中木質素的縮合程度低于稻草,球磨及再生對竹材木質素結構沒有影響。竹材原料球磨再生后的纖維結晶區有一定程度破壞,結晶度有所下降。竹材無論是化學成分還是生物結構均與木材類似,在溶解性能上更接近于木材。基于LiCl/DMSO溶劑體系的木質素分離不會破壞竹材木質素的化學結構,同時有助于提高竹材木質素組分的分離效果。
猜你喜歡
哲學評論(2021年2期)2021-08-22 01:53:34
新世紀智能(數學備考)(2020年11期)2021-01-04 00:38:16
中華詩詞(2019年7期)2019-11-25 01:43:04
中國外匯(2019年17期)2019-11-16 09:31:14
模具制造(2019年3期)2019-06-06 02:10:54
影視與戲劇評論(2016年0期)2016-11-23 05:26:01
現代企業(2015年9期)2015-02-28 18:56:50
現代企業(2015年1期)2015-02-28 18:43:18
新高考·高一物理(2014年1期)2014-09-18 01:26:07
土木建筑工程信息技術(2013年2期)2013-10-17 03:14:12
