CT和MRI檢查小兒骨肌原始神經外胚層腫瘤的臨床分析
2020-09-17 11:54:16謝浩勛羅圣熙
影像研究與醫學應用 2020年19期
謝浩勛,羅圣熙
(1廣州市社會福利院放射科 廣東 廣州 510520)
(2廣州市白云區人民醫院神經內科 廣東 廣州 510500)
作為一種極為罕見的臨床疾病,起源于神經外胚層的原始神經外胚層腫瘤產生的根本原因在于腫瘤原始的小圓形細胞沒有分化,繼而形成高度惡性腫瘤。根據類別差異總體可以劃分為兩種類型,即中樞性和外周性[1]。在此過程中產生的外周性原始神經外胚層腫瘤主要產生了外周神經系統,本疾病更多發生于兒童和青少年,在影像學中的展現也具有多樣性的特征,自身并沒有較為顯著的發病癥狀。
1 資料與方法
1.1 一般資料
選取2013年1月—2019年12月間廣州市白云區人民醫院收治的外周性原始神經外胚層腫瘤患兒20例,其中男性11例,女性9例,年齡2~12歲,平均年齡7歲,患兒病程1個月~6個月不等,平均3.5個月,患者臨床表現為有局部疼痛感,頜面部發生病變引發視力和嗅覺能力降低,胸悶咳嗽,軟組織發現組織腫塊等。
1.2 掃描方法及參數
在CT掃描過程中主要采用 CT750 HD螺旋CT機,掃描參數:選用120kV,電流選用自動管電流調節,螺距為1.375,層厚和層間距均為5mm,重建間隔為0.625mm。掃描時選用碘普羅胺對比劑,劑量為1.5mL/kg,通過高壓注射器進行靜脈快速注射,注射速率在0.8~2.5mL/s之間。掃描工作完成后將所得數據上傳至工作站,進行多平面圖像重組,最大密度投影,容積再現等圖像處理技術對所得圖像進行后期處理。
MRI檢查:使用飛利浦公司3.OT型超導MR機,每層的距離為1mm,厚度為5mm。通過FLAIR序列做出T1WI(TR 550ms,TE 6.3ms),T2 WI(TR 4300ms,TE 110ms),T1-FLAIR(TR 2200ms、TI 860ms、TE 15ms),STIR T2 WI(TR 4000ms,TI 210ms、TE 60ms),對病變部位進行掃描,完成此過程后需要對患者體內注入Gd-DTPA(釓噴替酸葡甲胺),用量為0.1mmol/kg。然后再次對病變位置進行掃描,掃描的過程中基本參數保持不變。
2 結果
2.1 發病部位
在臨床治療過程中的20例腫瘤患者,起源于骨骼系統和軟組織的分別為10例,病變部位在肋骨的2例,肩腳骨和領骨以及作恥骨疾病為1例。而處于胸壁肌肉間隙中以及走側肩腳骨旁邊下肌等多個部位的腫瘤患者分別為2例。
2.2 影像學表現
在骨來源的10例疾病中,有6名男性,4名女性,而年齡處于3~9歲之間。通過CT檢查發現,CT表現為骨質破壞并存在周圍軟組織增厚的問題,病變的最大直接達到了2.5~7.5厘米,而骨質破壞的現象主要是溶骨性破壞,在5例臨床病例中,有2例患者存在瘤骨現象,在此周圍也已經展現出了混雜密度或者軟組織的基本密度,經過CT進行掃描之后發現其自身的周圍軟組織出現了強化的不均勻。
在軟組織來源的10例患者中,有3名男性,7名女性,其年齡為2~11歲。在所有患者中,有5名患者進行了CT例行檢查,而5例進行的使CT增強檢查。檢查之后的結果發現有5例患者的病變展現是軟組織密度略低,并且患者病變部位存在少量小片狀的鈣化現象。通過對兩例患者進行增強掃描發現,病變都存在強化不均勻現象,其自身的最大徑線達到了4.5~14.5厘米。
2.3 預后
針對本組20例患者都進行了手術切除,其中16例先進行了化療治療,之后才采取了手術治療的方式,而4例患者依然在化療治療。有3例患者在發病時已經發現病灶出現轉移,但是經過化療之后病灶已經消失。經過手術治療之后,發現患者在之后并沒有出現病情反復和轉移的問題。
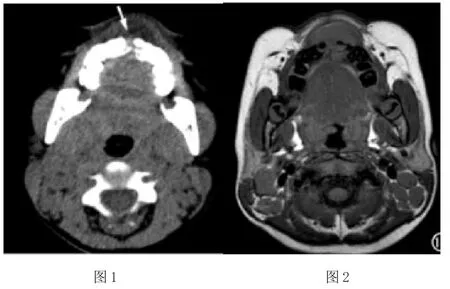
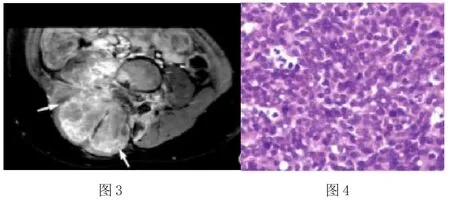
圖1CT平掃軟組織窗,顯示上頜骨中線有腫塊;圖2MRT1WI顯示上頜骨中線附近有低信號軟組織腫塊;圖3腰背部橫軸面增強掃描,顯示病變部位呈現明顯不均勻強化;圖4鏡下顯示腫瘤組織的組成,課件細胞核分裂。
3 討論
目前針對pPNETs自身的基本來源還沒有形成較為明確的報道和文獻記載,正是受到某一不確定因素的影響,繼而使得原始神經細胞和始基種子細胞等多種細胞產生突變和分化以及去分化現象,最終使得其向著神經上皮甚至間葉組織分化,最終形成腫瘤[2]。對于pPNETs來說,其自身屬于一種極為罕見的疾病,更會在身體的所有部位出現。在本組患兒中的年齡中分位是9歲,其中男性超過女性,其自身的發病原理和發病部位和當前的相關文獻記載較為符合[3]。
在組織上學上認為,pPNETs和其他小細胞腫瘤較為相似,在實際診斷過程中難以準確判斷,必須在在相關專業設備作用下完成多種神經分化情況的判斷,該種方式也是確診該疾病目前為止唯一的一種方式,根據世界衛生組織針對該種疾病的分類,認為Ewing肉瘤he PNET很難判斷和分別。更有作者認為兩者之所以難以辨別是因為兩者都能夠對原癌基因中的CD99進行表達[4]。在電鏡之下,pPNETs所展現出的使原始小圓細胞,其自身的特點是核濃染,標本中也可能會有壞死的病灶。在免疫組化檢查過程中,可見的CD99展現為陽性表達。該種數據和相關的文獻記載和報道相符合[5]。
對于pPNETs來說,其自身的影像學表現具有多樣性,本文主要對本組案例中的資料進行總結并對相關文獻進行復習,總體總結其自身的特點包括以下幾種:一是骨來源pPNETs自身具有骨質破壞性,在此過程中溶骨性破壞居多,形成瘤骨的可能性比較大,且伴有軟組織性包塊。此次研究中所發現的包塊直徑為3~8厘米,腫塊整體較大,在周圍組織結構中存在壓迫性物質,腫塊邊界周圍存在明顯的模糊性。二是在進行CT掃描的過程中可能會出現顯示不均勻的現象,偶爾還會出現鈣化現象,周圍的孤星結構會受到病變內囊的侵襲,繼而出現累骨內的T1和T2信號。三是在脊柱周圍,經過椎間孔可以實現pPNETs的進入,繼而對硬膜囊形成壓迫作用,在本組中產生的2例病變都發生在脊柱旁邊的軟組織。
對于pPNETs疾病來說,患者自身的預后恢復情況較差,通過對相關文獻的報道研究可以發現,超過70%的患者在疾病確診之后三年內出現死亡。而遠處轉移也較多出現在骨頭重,其次是肺部。在本組研究中,有兩例患者在發病的同時都出現了肺部轉移的現象,經過化療處理后病灶部分消失,只有1例患者在手術后兩年出現了腫瘤原位復發并且伴有肺部轉移的現象。
因為pPNETs在臨床醫學中并不多見,其自身的影像學展現也缺乏特異性,其自身和掐骨骼來源以及軟組織哀怨的惡性腫瘤較為相似,難以判斷和辨別,對于該疾病的確診過分依賴病理學技術。但是在臨床治療過程中,必須對本疾病進行考慮和分析,倘若質疑病變組織對神經系統形成了侵擾作用,而MRI對腫瘤邊界以及周圍神經結構的基本關系形成準確判斷,在手術開展之前進行必要的等級評定,形成詳細的手術開展計劃,對腫瘤復發和轉移情況進行評判。
