吡草醚懸浮劑和微乳劑的高效液相色譜分析方法
2020-09-11 08:38:04原萬玲馬杜康趙鵬躍黃啟良中國農業科學院植物保護研究所北京100193
現代農藥 2020年4期
關鍵詞:懸浮劑
原萬玲,馬杜康,孟 璨,趙鵬躍,黃啟良(中國農業科學院植物保護研究所,北京 100193)
吡草醚(pyraflufen-ethyl),化學名2-氯-5-(4-氯-5-二氟甲氧基-1-甲基吡唑-3-基)-4-氟苯氧基乙酸乙酯,商品名速草靈、丹妙藥、吡氟苯草酯等,由日本Nohyaku公司開發,是一種新型的苯基吡唑類苗后觸殺型除草劑,其作用機理是抑制植物體內的原卟啉Ⅳ氧化酶,利用小麥與雜草對藥劑吸收和代謝的差異,達到選擇性地防治小麥田闊葉雜草的效果[1-2],也可有效促使成熟期的棉花脫葉[3]。2013年國內企業首次登記吡草醚產品。目前,我國登記的含有吡草醚有效成分的農藥產品共9個,其中原藥3個,母藥1個,懸浮劑3個,微乳劑2個,應用于棉花和小麥田。然而,吡草醚農藥產品尚無國家或行業標準,不能依法進行檢驗檢測。因此,開展吡草醚農藥產品中有效成分測定通用分析方法的研究,可以加快制定標準的步伐,盡早解決依法檢驗檢測和依法監督管理問題。
有文獻報道了以鄰苯二甲酸二環己酯為內標物,利用氣相色譜分析吡草醚原藥的方法,該方法具有較好的線性、精密度和準確度[4]。劉同金等[5]采用氣相色譜建立了吡草醚在小麥及土壤中的殘留分析方法。秦旭等[6]采用高效液相色譜法測定了棉花中吡草醚的殘留。張煜卓等[7]采用QuEChERS技術與氣相色譜-串聯質譜法測定了南國梨中吡草醚的殘留。目前關于吡草醚制劑產品的分析方法尚未見報道。本文建立了在同一液相條件下測定吡草醚懸浮劑和微乳劑的方法,為吡草醚農藥制劑產品質量檢測提供依據。
1 材料與方法
1.1 儀器與試劑
Agilent 1260-DAD高效液相色譜儀(自動進樣器),Chemstation工作站,美國Agilent公司;色譜柱:Agilent ZORBAX SB-C18不銹鋼柱(250 mm×4.6 mm,5 μm);KQ3200B超聲波清洗器,昆山市超聲儀器有限公司;有機過濾器(微膜孔徑0.45 μm)。
乙腈(色譜純);二次蒸餾水;吡草醚標準品(質量分數99.97%),德國Dr.Ehrenstorfer公司;2%吡草醚懸浮劑,山東先達農化股份有限公司;2%吡草醚微乳劑,江蘇龍燈化學有限公司。
1.2 液相色譜操作條件
流動相:乙腈+水(體積比65∶35);柱溫:30℃;流速:1.0 mL/min;檢測波長:243 nm;進樣體積:5 μL;保留時間:吡草醚約9.5 min。
上述液相色譜操作條件是典型的操作參數,可以根據不同儀器特點和溫度條件,對給定的操作參數進行適當的調整,以獲得最佳效果。典型的吡草醚標準品、2%吡草醚懸浮劑、2%吡草醚微乳劑樣品高效液相色譜圖見圖1、2、3。
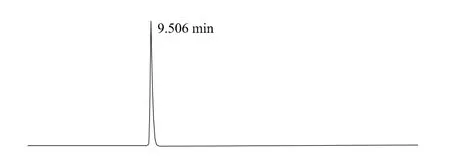
圖1 吡草醚標準品高效液相色譜圖
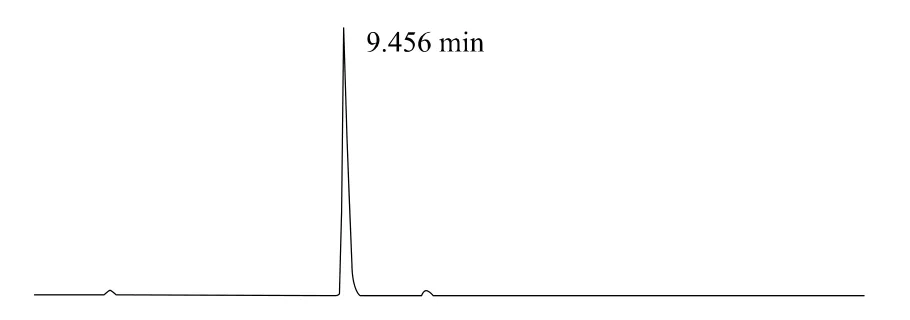
圖2 2%吡草醚懸浮劑高效液相色譜圖
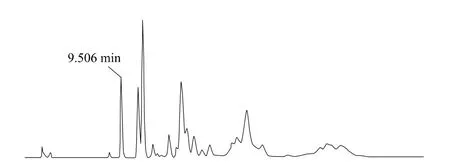
圖3 2%吡草醚微乳劑高效液相色譜圖
1.3 測定步驟
1.3.1 標準樣品的配制
稱取0.025 g(精確至0.000 01 g)吡草醚標樣于100 mL容量瓶中,加入20 mL甲醇,超聲波振蕩5 min,冷卻至室溫,用甲醇稀釋至刻度,搖勻,記為標樣溶液。
1.3.2 試樣溶液的配制
分別稱取2%吡草醚懸浮劑和2%吡草醚微乳劑試樣0.3 g(精確至0.000 01 g)于100 mL容量瓶中,加入20 mL甲醇,超聲波振蕩5 min,冷卻至室溫,用甲醇稀釋至刻度,搖勻,過濾待測。
1.3.3 測定
在上述操作條件下,待儀器穩定后,連續注入數針標樣溶液,直至相鄰兩針吡草醚峰面積相對變化小于1.2%后,按照標樣溶液、試樣溶液、試樣溶液、標樣溶液的順序進行測定。
1.3.4 計算
將測得的兩針試樣溶液以及試樣前后兩針標樣溶液中吡草醚峰面積分別進行平均。試樣中吡草醚質量分數w(%)按式(1)計算:

式中:w為試樣中吡草醚質量分數,%;A2為試樣溶液中吡草醚峰面積的平均值,mAU·s;m1為吡草醚標樣的質量,g;P為標樣中吡草醚質量分數,%;A1為標樣溶液中吡草醚峰面積的平均值,mAU·s;m2為試樣的質量,g。
2 結果與討論
2.1 流動相的選擇
由于2%吡草醚微乳劑中干擾較大,為使有效成分得到良好的分離效果,并且分析時間相對合適,峰形尖銳,保留時間適中,根據吡草醚的結構和性質,對不同配比的乙腈和水、甲醇和水流動相進行篩選。結果表明,流動相為乙腈+水(體積比65∶35),流速為1.0 mL/min,吡草醚保留時間為9.5 min,能夠獲得良好的分離效果。
2.2 檢測波長的選擇
利用紫外-可見檢測器對吡草醚溶液在225~380 nm范圍內進行掃描。吡草醚的紫外光譜圖如圖4所示。從圖4中可見,吡草醚的最大吸收波長約為243 nm,在該波長處靈敏度較高,各種雜質不影響吡草醚的測定,能夠滿足分析的要求,故將檢測波長確定為243 nm。
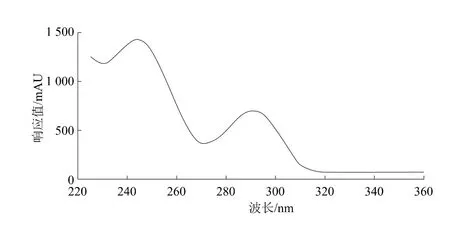
圖4 吡草醚紫外吸收譜圖
2.3 分析方法的特異性
本試驗采用HPLC-DAD峰純度分析法來鑒別吡草醚。吡草醚標樣、2%吡草醚懸浮劑、2%吡草醚微乳劑中的吡草醚HPLC-DAD峰純度均大于990,有效成分處無其他物質干擾,符合定量分析要求。
2.4 分析方法的線性關系
按1.3.1標準樣品的配制方法配制標樣溶液,用甲醇以1∶1的比例梯度稀釋四次,獲得5個濃度的有效成分線性相關溶液。待儀器穩定后,在上述操作條件下,按照分別進同樣體積的標樣,進行測定,取兩次測定的平均結果。以吡草醚質量濃度為橫坐標,峰面積為縱坐標繪制標準曲線。從圖5可以看出,當吡草醚質量濃度在15.75 mg/L~251.92 mg/L之間(進樣體積5 μL),與相應的吡草醚峰面積之間呈現良好的線性關系,計算得回歸方程為y=8.732 8 x+9.456 2,相關系R2=0.999 9,完全可以滿足定量分析要求。
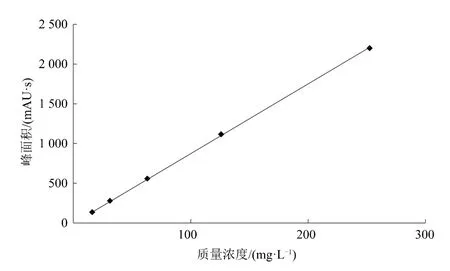
圖5 吡草醚標準曲線圖
2.5 分析方法的精密度
從同一樣品中稱取5個試樣,在上述色譜條件下進行分析,測得2%吡草醚懸浮劑和2%吡草醚微乳劑的標準偏差分別為0.07%和0.01%(表1)。
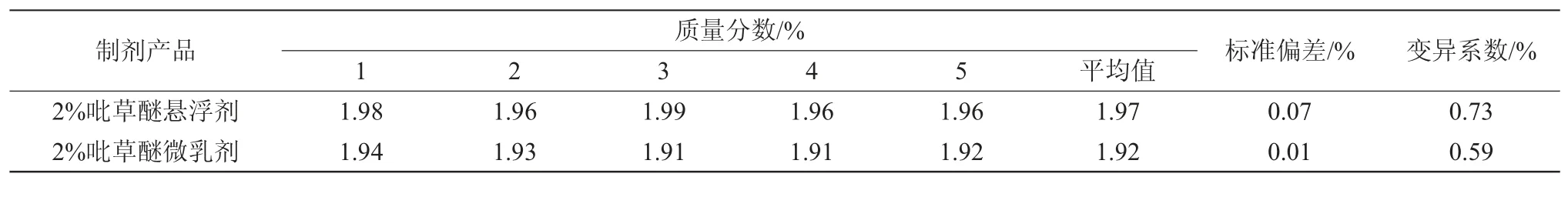
表1 分析方法的精密度試驗結果
2%吡草醚微乳劑中吡草醚質量分數測定結果的變異系數為0.59,小于修改的Horwitz公式2(1-0.51ogC)×0.67=2.43(其中C為樣品中有效成分質量分數的平均值),表明有效成分分析方法精密度的測定結果符合要求。
2%吡草醚懸浮劑中吡草醚質量分數測定結果的變異系數為0.73,小于修改的Horwitz公式2(1-0.51ogC)×0.67=2.42,表明有效成分分析方法精密度的測定結果符合要求。
2.6 分析方法的準確度
分別稱取5個已知含量的懸浮劑和微乳劑試樣于100 mL容量瓶中,加入1.3.1中配制的吡草醚標樣溶液4 mL,用甲醇超聲溶解,冷卻至室溫,用甲醇定容至刻度,搖勻。吡草醚的回收率按式(2)計算。
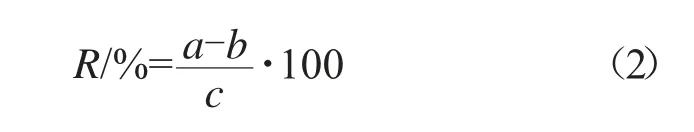
式中:R為回收率,%;a為測定量,mg;b為所稱原藥樣品中待測組分量,mg;c為理論加入量,mg。
經測定,2%吡草醚懸浮劑和2%吡草醚微乳劑的回收率分別為97.45%和97.68%,試驗結果見表2。表2表明有效成分分析方法準確度的測定結果符合要求。
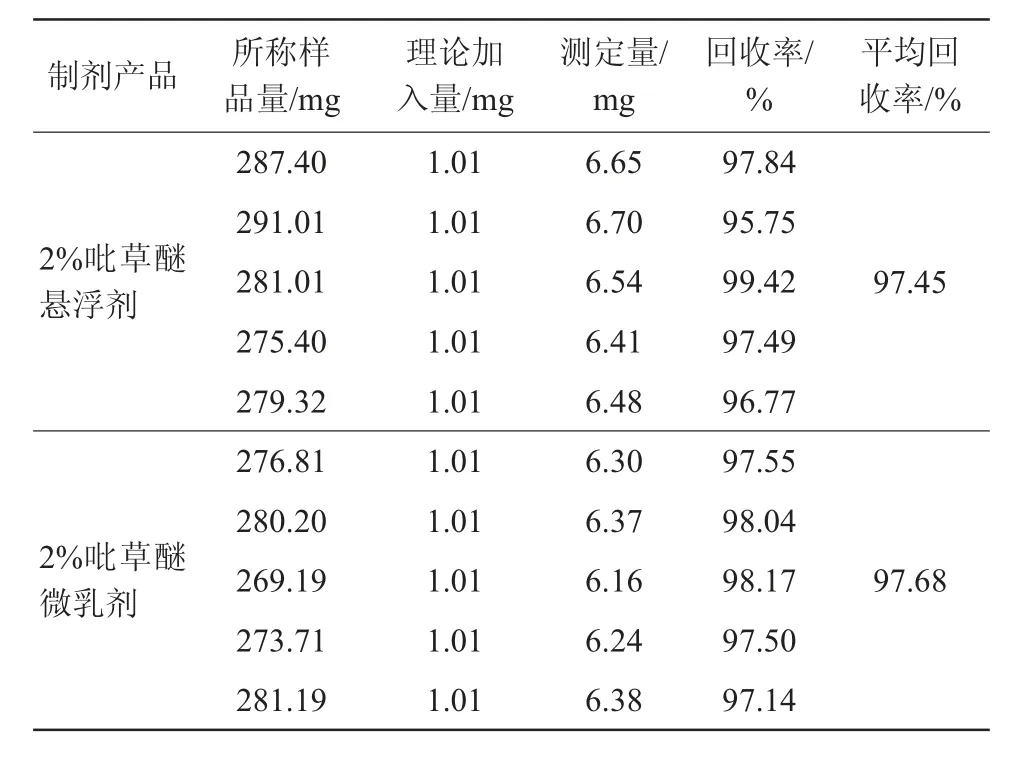
表2 分析方法的準確度試驗結果
3 結 論
利用高效液相色譜法對2%吡草醚懸浮劑和2%吡草醚微乳劑分別進行了定性和定量分析。結果表明,2%吡草醚懸浮劑和2%吡草醚微乳劑的回收率分別為97.45%和97.68%。所采用的高效液相色譜法準確度和精密度較高,線性關系較好,具有簡便、快速、準確及分離效果好等優點,能夠作為吡草醚制劑的分析測定方法。
猜你喜歡
鉆井液與完井液(2022年4期)2022-10-26 06:39:24
世界農藥(2019年4期)2019-12-30 06:25:10
農藥科學與管理(2019年7期)2019-11-29 07:35:14
農藥科學與管理(2019年9期)2019-11-23 08:41:08
農藥科學與管理(2019年8期)2019-11-23 08:04:44
農藥科學與管理(2019年5期)2019-08-13 00:47:58
農藥科學與管理(2019年10期)2019-04-20 07:13:10
現代園藝(2018年3期)2018-02-10 05:18:13
當代化工研究(2016年7期)2016-03-20 16:21:57
中國果菜(2016年9期)2016-03-01 01:28:41
