奧卡西平刻痕片仿制藥與原研藥溶出行為及相關指標的一致性評價研究
2020-07-01 01:52:58呂蓓蓓楊海源程華魏文芝張敏娟
中國藥房 2020年12期
呂蓓蓓 楊海源 程華 魏文芝 張敏娟

中圖分類號 R917 文獻標志碼 A 文章編號 1001-0408(2020)12-1463-08
DOI 10.6039/j.issn.1001-0408.2020.12.11
摘 要 目的:評價奧卡西平刻痕片仿制藥與原研藥溶出行為的一致性,并比較兩者的外觀、半片制劑的脆碎度及分割質量損失以及不同企業原料藥的晶型、晶體形貌。方法:采用高效液相色譜法測定含量;采用槳法(設置轉速60 r/min,溫度37.0 ℃)測定仿制藥與原研藥在4種不同溶出介質[含0.6%十二烷基硫酸鈉的鹽酸溶液(pH=1.2)、含0.6%十二烷基硫酸鈉的醋酸鹽緩沖溶液(pH=4.5)、含0.6%十二烷基硫酸鈉的磷酸鹽緩沖溶液(pH=6.8)、含0.6%十二烷基硫酸鈉的水溶液]中的累積溶出度,采用相似因子法評價兩者溶出曲線的相似性,并評價半片與整片制劑的批內均一性;采用脆碎度檢測儀及電子天平測定半片制劑的脆碎度及分割質量損失;采用X射線衍射儀及掃描電子顯微鏡觀察不同企業原料藥的晶型和晶體形貌。結果:奧卡西平檢測質量濃度的線性范圍為33.42~401.09 μg/mL(r=0.999 9);定量限為0.10 μg/mL,檢測限為0.04 μg/mL;精密度、穩定性、重復性、耐用性試驗的RSD均小于2%;回收率為99.80%~101.63%(RSD為0.37%~0.91%,n=3)。仿制藥A、B和原研藥在4種不同溶出介質中90 min時的平均累積溶出度分別為92%、87%、90%[含0.6%十二烷基硫酸鈉的鹽酸溶液(pH=1.2)];94%、94%、90%[含0.6%十二烷基硫酸鈉的醋酸鹽緩沖溶液(pH=4.5)];95%、95%、91%[含0.6%十二烷基硫酸鈉的磷酸鹽緩沖溶液(pH=6.8)];97%、98%、95%(含0.6%十二烷基硫酸鈉的水溶液)。仿制藥A、B和原研藥在上述4種溶出介質中的相似因子分別為66、81,71、69,71、61,59、39。前15 min時,仿制藥A、B和原研藥半片與整片的溶出度差值分別為-3%~13%、-2%~24%、-3%~7%;仿制藥A半片、整片累積溶出度的RSD分別為6%~14%、2%~9%(n=12),仿制藥B為4%~10%、1%~8%(n=12),原研藥為2%~7%、2%~8%(n=12)。原研藥外觀為梭形,刻痕較深;仿制藥片形各異,且刻痕明顯淺于原研藥。仿制藥A、B和原研藥的脆碎度、分割質量損失分別為0.62%、0.67%,0.12%、0.11%,0.08%、0.05%。國產原料藥呈不規則的塊狀和碎屑,原研藥企業產原料藥呈規則的扁平長方體和規則的條狀且碎屑少;但兩者的X射線衍射特征峰基本一致。結論:仿制藥A在4種溶出介質中的溶出行為與原研藥一致;仿制藥B在含0.6%十二烷基硫酸鈉的水溶液中的溶出行為與原研藥不同;原研藥掰分前后批內均一性無明顯變化,而仿制藥A、B掰分后的批內均一性較整片有所降低;仿制藥的脆碎度和分割質量損失均高于原研藥;兩者原料藥晶型相同但晶體形貌存有差異。
關鍵詞 奧卡西平;溶出行為;刻痕片;仿制藥;原研藥;一致性評價;高效液相色譜法
Consistency Evaluation on the Dissolution Behavior and Related Indicators between Generic and Original Preparations of Oxcarbazepine Scored Tablets
LYU Beibei1,2,YANG Haiyuan1,2,CHENG Hua1,2,WEI Wenzhi1,2,ZHANG Minjuan1,2(1. Chemistry Laboratory, Qinghai Institute for Drug Control, Xining 810016, China; 2. Qinghai Provincial Key Laboratory of Modernization of Traditional Chinese and Tibetan Medicine, Xining 810016, China)
ABSTRACT ? OBJECTIVE: To evaluate the dissolution behavior consistency between the generic drugs and original drugs of Oxcarbazepine scored tablets, and to compare the appearance, the friability of the split portions, loss of mass of the split portions as well as crystal form and morphology of raw material from different enterprises. METHODS: HPLC method was adopted. The paddle method (rotation speed of 60 r/min, the temperature of 37.0℃) was adopted to determine accumulative dissolution rate of generic and original drugs in 4 mediums [0.6% SDS hydrochloric acid solution (pH=1.2), 0.6% SDS acetate buffer solution (pH=4.5), 0.6% SDS phosphate buffer solution (pH=6.8) and 0.6% SDS water solution]. The similarity factor method was used to evaluate the similarity of dissolution curves as well as intra-batch uniformity of the split portions and whole tablets. The friability tester and electronic balance were used to determine the friability and the loss of mass of the split portions. X-ray diffractometer and scanning electron microscope were used to observe the crystal form and crystal morphology of the raw materials of different enterprises. RESULTS: The linear range of oxcarbazepine was 33.42-401.09 μg/mL(r=0.999 9);LOQ was 0.10 μg/mL, and LOD was 0.04 μg/mL; RSDs of precision,stability, reprodu- cibility and durability tests were lower than 2.0%; the reco- veries were 99.80%-101.63% (RSD=0.37%-0.91%,n=3). The average cumulative dissolution rate of generic drug A, generic drug B and original drug in 4 different dissolution media at 90 min were 92%, 87%, 90% [0.6% SDS hydrochloric acid solution (pH=1.2)];94%, 94%, 90% [0.6% SDS acetate buffer solution (pH=4.5)];95%, 95%, 91% [0.6% SDS phosphate buffer solution (pH 6.8)]; 97%, 98%, 95% (0.6% SDS water solution). The similarity factors of generic drug A, generic drug B and original drug in 4 kinds of different dissolution media were 66 and 81,71 and 69,71 and 61,59 and 39. In the first 15 min, the difference of dissolution rate of split portions and whole tablets were -3%-13%, -2%-24% and -3%-7% for generic drug A, generic drug B and original drug, respectively. RSDs of accumulative dissolution rate of split portions and whole tablets were 6%-14% and 2%-9% for generic drug A (n=12), 4%-10% and 1%-8% for generic drug B (n=12) and 2%-7% and 2%-8% for original drug. The appearance of the original drug was fusiform, and the notch was deep;the shape of the generic drug was different from each other, and the notch of the generic drug was significantly shallower than that of original drug. The friability, the loss of mass of the split portions for generic drug A and generic drug B, original drug were 0.62%and 0.67%,0.12% and 0.11%,0.08% and 0.05%. The domestic raw materials possessed irregular lumps and debris, while the raw materials produced by original drug enterprises possessed regular flat cuboids and regular strips with little debris; but X-ray diffraction peaks of them were basically the same. CONCLUSIONS: The dissolution behavior of generic drug A in 4 medium is consistent with that of the original drug; dissolution behavior of generic drug B in water containing 0.6%SDS is different from that of the original drug; there is no significant change in the homogeneity of the original drug before and after splitting, but the homogeneity of the generic drug A and B after splitting is lower than that of the whole tablet; the fragility of generic drugs and loss of mass of split portions are higher than those of the original drugs; two kinds of raw material have the same crystal form but different crystal morphology.
KEYWORDS ? Oxcarbazepine; Dissolution behavior; Scored tablets; Generic drug; Original drug; Consistency evaluation; HPLC
功能性刻痕片劑(可分割的刻痕片)是一種帶有一道或多道刻痕,以便于進行劑量分割的片劑[1]。其具有靈活調整藥物劑量、彌補兒童或老年用藥規格不足、降低患者用藥成本的特點,但存在分劑量不均勻[2-3]、分割時出現粉末或碎片導致藥量損失的現象[4]。分劑量不均勻會影響藥物的療效或引發不良反應,特別是對于治療窗窄的藥物[5-6]。9.0版《歐洲藥典》[7]、41版《美國藥典》[8]均有相應的關于刻痕片的技術要求和規范,而《中國藥典》暫未收錄相關內容。美國FDA于2013年發布了相關技術指南[9],進一步對刻痕片劑進行了管理和規范。我國國家藥品監督管理局藥品審評中心(CDE)網站于2019年發布了《仿制口服片劑功能性刻痕設計和研究的一般要求》[1],要求仿制藥的功能性刻痕應該與原研藥保持一致,且須對分割后的制劑單元進行質量差異或含量均勻度、分割質量損失、脆碎度、溶出度等進行考察。功能性刻痕片作為口服固體制劑,溶出度是其關鍵質控屬性之一[10]。采用溶出曲線測定原研藥與仿制藥的溶出度,既可為仿制藥研發過程中處方的篩選提供參考,又可作為仿制藥質量與療效一致性評價的重要手段[11]。
奧卡西平(Oxcarbazepine)是一種新型抗癲癇藥物,為卡馬西平的10-酮基衍生物,主要通過其活性代謝產物10-單羥基代謝物發揮作用,具有耐受性好、毒副作用小、藥物間相互作用少等特點,已成為治療成人及兒童癲癇局部發作或全身強直性陣攣發作的一線藥物[12]。奧卡西平刻痕片原研藥可掰分為兩個半片,極大地彌補了兒童用藥規格不足的情況[13]。
本研究通過高效液相色譜法(HPLC)結合相似因子法比較了不同溶出介質中奧卡西平仿制藥與原研藥的溶出曲線,并對掰分后的半片制劑進行了溶出曲線考察,以剖析奧卡西平刻痕片整片制劑與半片制劑的溶出行為;同時,本研究還比較了全片外觀,半片制劑的分割質量損失、脆碎度及原料藥的晶型、晶體形貌,旨在評價仿制藥與原研藥的一致性,并為仿制藥的生產工藝和內在質量提升提供參考,亦為刻痕片的質量控制提供依據。
1 材料
1.1 儀器
1260型HPLC儀,配備G7111A型四元泵、G7129A型自動進樣器、G7116A型柱溫箱、G71157型二極管陣列檢測器(美國Agilent公司);AT型全自動溶出儀(瑞士Sotax公司);FAVD-25型真空脫氣儀(上海富斯科分析儀器有限公司);PB-21型酸度計、CP225D型十萬分之一電子天平(德國Sartorious公司);H1850R型高速冷凍離心機(湖南長沙湘儀離心機儀器公司);D8 Advance型X射線衍射儀(德國Bruker公司);JSM-6390型掃描電子顯微鏡(日本JEOL公司);FT-2000型脆碎度檢測儀(天津市矽新科技有限公司);KQ-500DE型數控超聲波清洗器(昆山市超聲儀器有限公司)。
1.2 藥品與試劑
奧卡西平對照品(中國食品藥品檢定研究院,批號:100657-201102,純度:99.8%);奧卡西平刻痕片原研藥(瑞士Novartis公司,以下簡稱“企業R”,規格:300 mg,批號:TR228、T2506、TU899);仿制藥A(國內A企業,規格:300 mg,批號:180411、180109、190103);仿制藥B(國內B企業,規格:300 mg,批號:181109、190101、171003);奧卡西平原料藥1(國內企業1,批號:180602,純度:99.4%);奧卡西平原料藥2(國內企業2,批號:170620,純度:99.2%);奧卡西平原料藥3[北京諾華制藥有限公司(即原研藥企業中國分公司)贈送,批號:181105,純度:99.6%];十二烷基硫酸鈉(SDS)、乙腈均為色譜純,其余試劑均為分析純,水為超純水。
2 方法與結果
2.1 色譜條件
色譜柱:Thermo BDS Hypersil C18(150 mm×4.6 mm,5 μm);流動相:乙腈-0.05 mol/L磷酸二氫鉀水溶液(含0.2%三乙胺,用磷酸調節pH至6.0)(40 ∶ 60,V/V);檢測波長:256 nm;流速:1.0 mL/min;柱溫:30 ℃;進樣量:10 μL。
2.2 溶出介質的選擇與配制
在2015年版《中國藥典》(二部)[14]、美國FDA橙皮書數據庫[15]、41版《美國藥典》[8]收錄的奧卡西平片的質量標準中,均在溶出介質中加入了SDS(每300 mg的質量百分比均為0.6%)以提高藥物的溶解度。為考察溶液pH對奧卡西平溶解性能的影響,本研究分別測定奧卡西平原料藥(批號:180602)在含0.6%SDS的鹽酸溶液(pH=1.0、pH=2.0)、含0.6%SDS的醋酸鹽緩沖溶液(pH=4.5)、含0.6%SDS的磷酸鹽緩沖溶液(pH=6.0、pH=6.8)、含0.6%SDS的水溶液中的溶解度。結果發現,奧卡西平在不同pH條件下的溶解度均在0.3 mg/mL左右,說明pH對奧卡西平溶解度無明顯影響。根據《普通口服固體制劑溶出度試驗技術指導原則》[16],本研究最終選擇了含0.6%SDS的鹽酸溶液(pH=1.2)、含0.6%SDS的醋酸鹽緩沖溶液(pH=4.5)、含0.6%SDS的磷酸鹽緩沖溶液(pH=6.8)、含0.6%SDS的水溶液為溶出介質,并參照《普通口服固體制劑溶出曲線測定與比較指導原則》[17]中溶出介質配制方法進行配制。
2.3 溶液的制備
2.3.1 對照品溶液 精密稱取奧卡西平對照品33 mg,置于100 mL量瓶中,加乙腈10 mL,超聲(功率:500 W,頻率:40 kHz,下同)使溶解,分別用4種溶出介質稀釋至刻度,即得4種質量濃度均為330 μg/mL的對照品溶液。
2.3.2 供試品溶液 ①按2015年版《中國藥典》(四部)通則“0931第二法(槳法)”[18]分別取“2.2”項下4種溶出介質900 mL,置于溶出杯中。取9批樣品,每批12片,分別置于上述溶出杯中,設置溫度37.0 ℃、轉速60 r/min,分別于5、10、15、20、30、45、60、90 min時取溶液5 mL,再經自動溶出儀自動補加相應溶出介質5 mL;取樣溶液經0.45 μm濾膜濾過,取續濾液,即得。②取樣品適量,精密稱定,置于500 mL量瓶中,加入含0.6%SDS的水溶液適量,振搖45 min,用含0.6%SDS的水溶液稀釋至刻度,搖勻,濾過,取續濾液,即得。③取樣品適量,精密稱定,分別置于100 mL量瓶中,加乙腈10 mL,超聲使溶解,用含0.6%SDS的水溶液稀釋至刻度,搖勻,濾過,取續濾液,即得。
2.3.3 空白對照溶液 參照輔料成分較多的仿制藥A的處方工藝,按比例稱取各輔料適量,混勻,加入含0.6%SDS的水溶液900 mL,超聲10 min,經0.45 μm濾膜濾過,取續濾液,作為空白對照溶液。
2.4 含量測定
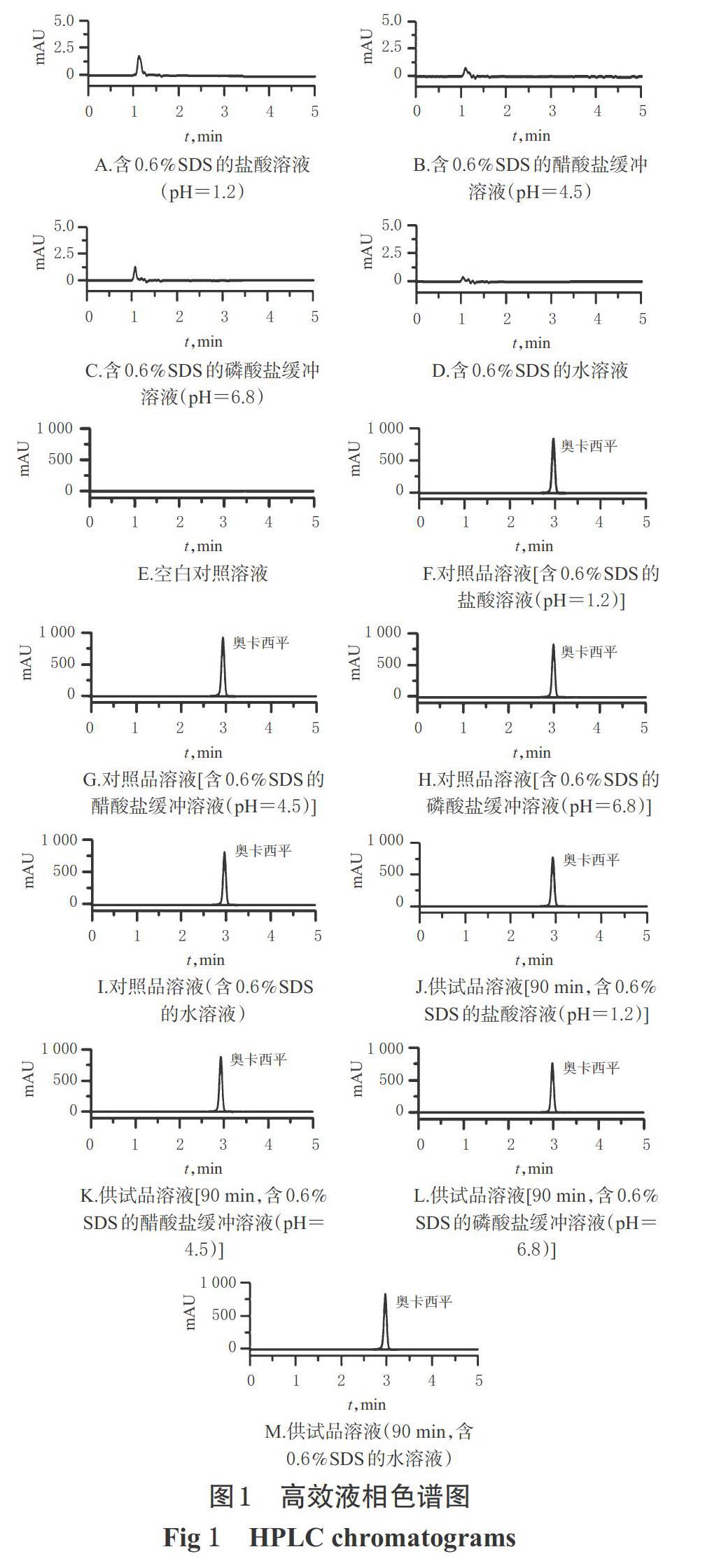
2.4.1 專屬性試驗 取“2.2”項下4種溶出介質、“2.3.3”項下空白對照溶液、“2.3.1”項下4種對照品溶液及“2.3.2①”項下4種供試品溶液各適量,按“2.1”項下色譜條件進樣測定,記錄色譜圖。結果顯示,4種溶出介質和空白對照對測定均無干擾,奧卡西平和相鄰雜質的分離度均大于1.5,理論板數按奧卡西平計均不低于3 000,詳見圖1。
2.4.2 線性關系考察 精密稱取奧卡西平對照品83.56 mg,置于50 mL量瓶中,加乙腈30 mL,超聲使溶解,加水稀釋至刻度,得質量濃度為1 671.20 μg/mL的對照品貯備液。分別精密量取上述對照品貯備液0.5、1.0、2.0、3.0、4.0、5.0、6.0 mL,置于25 mL量瓶中,用含0.6%SDS的水溶液稀釋至刻度,搖勻,制得系列線性對照品溶液,按“2.1”項下色譜條件進樣測定,記錄峰面積。以待測成分質量濃度(x,μg/mL)為橫坐標、峰面積(y)為縱坐標進行線性回歸,得奧卡西平的回歸方程為y=16.182x+30.237(r=0.999 9)。這表明,奧卡西平檢測質量濃度的線性范圍為33.42~401.09 μg/mL。
2.4.3 定量限與檢測限考察 參考預試驗結果取“2.4.2”項下質量濃度為33.42 μg/mL的線性對照品溶液,精密量取1.0 mL,置于100 mL量瓶中,用含0.6%SDS的水溶液稀釋至刻度,搖勻,精密量取3.0 mL,置于10 mL量瓶中,用含0.6%SDS的水溶液稀釋至刻度,搖勻,按“2.1”項下色譜條件進樣測定,以信噪比為10 ∶ 1計算得奧卡西平的定量限為0.10 μg/mL;取上述信噪比為10 ∶ 1的對照品溶液,精密量取4.0 mL,置于10 mL量瓶中,用含0.6%SDS的水溶液稀釋至刻度,搖勻,按“2.1”項下色譜條件進樣測定,以信噪比為3 ∶ 1計算得奧卡西平的檢測限為0.04 μg/mL。
2.4.4 精密度試驗 取“2.3.1”項下對照品溶液(以0.6%SDS的水溶液為溶劑),按“2.1”項下色譜條件連續進樣測定6次,記錄峰面積。結果,奧卡西平峰面積的RSD為0.2%(n=6),表明儀器精密度良好。
2.4.5 穩定性試驗 取“2.3.2①”項下溶出90 min時的4種供試品溶液各適量(仿制藥A,批號:180109),分別于室溫下放置0、2、4、6、8、10、12、14、16、18、20、24 h時,按“2.1”項下色譜條件進樣測定,記錄峰面積。結果,當以含0.6%SDS的鹽酸溶液(pH=1.2)、含0.6%SDS的醋酸鹽緩沖溶液(pH=4.5)為溶出介質時,供試品溶液放置10 h內峰面積的RSD分別為1.9%、1.7%(n=6),12~24 h內峰面積的RSD分別為2.2%、2.4%(n=7);當以含0.6%SDS的磷酸鹽緩沖溶液(pH=6.8)、含0.6%SDS的水溶液為溶出介質時,供試品溶液放置24 h內峰面積的RSD分別為0.5%、0.4%(n=12)。這表明,當以含0.6%SDS的鹽酸溶液(pH=1.2)、含0.6%SDS的醋酸鹽緩沖溶液(pH=4.5)為溶出介質時,供試品溶液放置10 h內穩定性良好,當以含0.6%SDS的磷酸鹽緩沖溶液(pH=6.8)、含0.6%SDS的水溶液為溶出介質時,供試品溶液放置24 h內穩定性良好。
2.4.6 重復性試驗 取仿制藥A(批號:180109)粉碎,取細粉約200 mg(約相當于奧卡西平150 mg),共6份,按“2.3.2②”項下方法制備供試品溶液,再按“2.1”項下色譜條件進樣測定,記錄峰面積并按外標法計算樣品中奧卡西平的含量。結果,奧卡西平平均含量的RSD為1.2%(n=6),表明方法重復性良好。
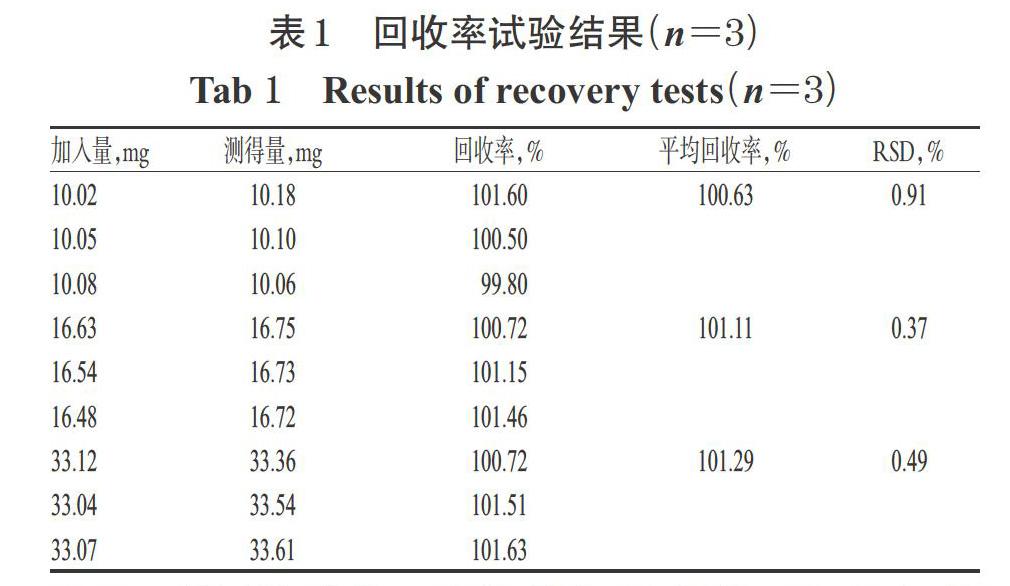
2.4.7 回收率試驗 按仿制藥A處方量的30%、50%、100%分別精密稱取奧卡西平對照品適量,并按其處方工藝稱取輔料,共9份,分別置于100 mL量瓶中,加乙腈10 mL,超聲使溶解,用含0.6%SDS的水溶液稀釋至刻度,搖勻,按“2.1”項下色譜條件進樣測定,記錄峰面積并計算回收率,結果見表1。
2.4.8 耐用性試驗 取仿制藥A(批號:180109),按“2.3.2①”項下方法制備供試品溶液(90 min),再按“2.1”項下色譜條件,分別以不同色譜住[Agilent Zorbax SB C18(150 mm×4.6 mm,5 μm)、Waters SunFire C18(150 mm×4.6 mm,5 μm)、Thermo BDS Hypersil C18(150 mm×4.6 mm,5 μm)]、HPLC儀(e2695型、1260型、LC-20AT型)、柱溫(25、30、35 ℃)]進樣測定,記錄峰面積并按外標法計算樣品含量。結果,奧卡西平含量的RSD均小于2.0%(n=3),表明該方法能夠滿足試驗要求,耐用性良好。
2.4.9 樣品含量測定 取9批樣品細粉,每份約40 mg(約相當于奧卡西平30 mg),按“2.3.2③”項下方法制備供試品溶液,再按“2.1”項下色譜條件進樣測定,記錄峰面積并按外標法計算樣品含量,平行操作2次。結果,仿制藥A、B及原研藥的平均含量分別為100.2%、99.9%、99.6%。
2.5 濾膜吸附性考察
以0.6%SDS的水溶液為溶出介質,取仿制藥A(批號:180109)適量,按“2.3.2①”項下方法制備供試品溶液,取90 min時的供試品溶液150 mL,置于500 mL量瓶中,用含0.6%SDS的水溶液稀釋至刻度(約相當于第1個取樣時間點奧卡西平的質量濃度),搖勻,作為濾膜吸附性考察溶液。取上述濾膜吸附性考察溶液適量,以 4 000 r/min離心10 min,取上清液,作為離心樣品;另取濾膜吸附性考察溶液,分別用濾膜(0.45 μm)依次濾過3、5、7、10 mL,作為濾膜濾過樣品。同時為考察自動溶出儀取樣管路是否存在吸附性,分別采用手動取樣和儀器自動取樣的方式取樣,按上述方法分別濾過5、10 mL濾膜吸附性考察溶液,作為手動取樣濾過樣品和自動取樣管路濾過樣品。取上述各樣品適量,分別按“2.1”項下色譜條件進樣測定,記錄峰面積并計算吸附率:吸附率(%)=(1-濾過樣品峰面積/離心樣品峰面積)×100%。結果,3、5、7、10 mL濾膜濾過樣品的吸附率分別為0.7%、0.3%、0.5%、0.2%,5、10 mL手動濾過樣品的吸附率分別為0.2%、0.3%,5、10 mL自動取樣吸附率分別為0.4%、0.1%(均小于2.0%),表明濾膜、自動取樣管路對樣品均無吸附性。
2.6 溶出行為考察
2.6.1 仿制藥與原研藥(整片)的溶出度 取9批樣品,每批12片,按“2.3.2①”項下方法考察其在4種溶出介質中的溶出行為,并以溶出時間為橫坐標(x)、各時間點樣品累積溶出度的平均值為縱坐標(y),繪制溶出曲線[累積溶出度(%)=An+[(An-1+…A2+A1)×V1
V2] ,式中V1為各時間點固定取樣體積,V2為溶出介質體積,其中An為各時間點測得溶出度[19]]。結果,仿制藥與原研藥整片制劑的溶出曲線在以含0.6%SDS的水溶液中存在較大差異,其中原研藥和仿制藥A、B在10 min時的累積溶出度分別為77%、66%、48%,說明原研藥在該時間點的溶出速度明顯快于仿制藥。原研藥和仿制藥A、B在其余3種溶出介質中10 min時的累積溶出度分別為68%、65%、64%[含0.6%SDS的鹽酸溶液(pH=1.2)];71%、64%、69%[含0.6%SDS的醋酸鹽緩沖溶液(pH=4.5)];72%、65%、62%[含0.6%SDS的磷酸鹽緩沖溶液(pH=6.8)],提示3種樣品在其余3種溶出介質中的溶出行為無明顯差異。原研藥和仿制藥A、B在含0.6%SDS的鹽酸溶液(pH=1.2)中累積溶出度超過85%所需的時間分別為45、30、45 min;在其余3種溶出介質中,不同企業樣品的累積溶出度在30 min時均達到85%以上。仿制藥A、B和原研藥在4種不同溶出介質中90 min時的平均累積溶出度分別為92%、87%、90%[含0.6%SDS的鹽酸溶液(pH=1.2)],94%、94%、90%[含0.6%SDS的醋酸鹽緩沖溶液(pH=4.5)],95%、95%、91%[含0.6%SDS的磷酸鹽緩沖溶液(pH=6.8)],97%、98%、95%(含0.6%SDS的水溶液),詳見圖2。
式中Rt為t時點原研藥的平均累積溶出度,Tt為t時點仿制藥的平均累積溶出度,n為取樣時點的個數[17]。結果顯示,仿制藥A、B與原研藥在含0.6%SDS的鹽酸溶液(pH=1.2)、含0.6%SDS的醋酸鹽緩沖溶液(pH=4.5)、含0.6%SDS的磷酸鹽緩沖溶液(pH=6.8)、含0.6%SDS的水溶液中的相似因子分別為66、81,71、69,71、61,59、39。表明仿制藥A在4種溶出介質中的溶出曲線與原研藥相似,而仿制藥B在含0.6%SDS的水溶液中的溶出曲線與原研藥存在差異,提示含0.6%SDS的水溶液具有區分力。
2.6.2 仿制藥與原研藥(半片)的溶出度及其差異 根據國家CDE網站發布的《仿制口服片功能性刻痕設計和研究的一般要求》,刻痕片分割后的部分的溶出度應符合成品質量標準[1]。2015年版《中國藥典》(二部)中記載奧卡西平片在含0.6%SDS的水溶液中30 min時的溶出度應不低于75%,因此掰分后的半片制劑在含0.3%SDS的水溶液中30 min時的溶出度也應不低于75%[14]。本研究按“2.6.1”項下方法考察了半片制劑的溶出行為。結果,仿制藥A、B及原研藥掰分后的半片制劑在上述條件下的溶出度分別為95%、93%、95%,均符合要求。為進一步考察奧卡西平刻痕片掰分后的溶出行為變化,取9批樣品,每批12片,掰成半片,考察其在4種溶出介質中與整片制劑的溶出行為差異。結果,前15 min內,仿制藥A、B的半片制劑在4種溶出介質中的溶出度均顯著升高,較原研藥差異明顯,其中仿制藥B半片與整片制劑的溶出度差值范圍最大,為-2%~24%,仿制藥A為-3%~13%,原研藥為-3%~7%,詳見圖3(因180401、190101、TR228批樣品的半片制劑在5 min時溶出度的RSD超過20%,故剔除5 min時間點的數據,下同)。
2.6.3 半片與整片制劑的批內均一性 為考察不同企業樣品掰分后的溶出均一性變化,本研究按2015年版《中國藥典》(二部)中奧卡西平片的溶出介質(整片制劑為含0.6%SDS的水溶液,半片制劑為含0.3%SDS的水溶液),隨機選取不同企業的1批樣品(仿制藥A批號:180109、仿制藥B批號:190101、原研藥批號:TU899),每批12片,按“2.3.2①”項下方法制備供試品溶液,以累積溶出度的RSD為指標,考察同一批樣品半片與整片的批內均一性(剔除5 min時間點)。結果,仿制藥A半片與整片各時間點累積溶出度的RSD分別為6%~14%、2%~9%(n=12),仿制藥B為4%~10%、1%~8%(n=12),原研藥為2%~7%、2%~8%(n=12)。這表明,原研藥掰分前后批內均一性無明顯變化,而仿制藥A、B掰分后批內均一性較整片有所降低。
2.7 外觀
奧卡西平原研藥外觀為梭形,刻痕較深;仿制藥片形各異,呈圓形或膠囊型,刻痕明顯淺于原研藥。采用手掰的方式進行分割后發現,仿制藥掰分時比較費力且掰分后不均勻;原研藥易于操作,掰分后均勻整齊。
2.8 脆碎度及分割質量損失考察
取9批樣品,每批12片,采用脆碎度檢測儀及十萬分之一電子天平測定掰分后半片制劑的脆碎度和分割質量損失。結果,原研藥的平均脆碎度、分割質量損失分別為0.08%、0.05%,仿制藥A分別為0.62%、0.67%,仿制藥B分別為0.12%、0.11%,均符合相關要求[1],但仿制藥的脆碎度和分割質量損失均高于原研藥。
2.9 原料藥的晶型與晶體形貌比較
有文獻報道,奧卡西平粉末X射線衍射的特征峰位于7.236°、10.220°、11.977°、14.399°、17.700°、19.098°、19.457°、20.060°、21.120°、21.978°、23.040°、23.658°、25.200°、25.637°、26.141°處[20]。本研究參照上述文獻對不同企業奧卡西平原料藥(原料藥1~3)進行X射線衍射測定。結果,3種原料藥X射線衍射圖譜幾乎完全相同,特征峰與文獻報道[20]基本一致,表明3種原料藥晶型一致(圖4)。將3種原料藥進行電鏡掃描發現,原料藥1、2的晶體形貌呈不規則的塊狀和碎屑,原料藥3呈規則的扁平長方體和規則的條狀且碎屑少(圖5)。這表明,國產原料藥與原研藥企業產原料藥的晶體形貌有所不同。
3 討論
2015年版《中國藥典》(二部)中以乙腈-0.05 mol/L磷酸二氫鉀水溶液(用磷酸調節pH至3.0)(40 ∶ 60,V/V)為流動相[14]。但筆者在試驗過程中發現,當水相pH為3.0時,對照品溶液在緊鄰主峰前出現一小峰,無法基線分離;且在調整流動相比例以及更換不同色譜柱后均無明顯改善。但調整水相的pH(pH=4.0、5.0、6.0)且當其pH為6.0時,該小峰消失,更換不同色譜柱以及不同品牌HPLC儀后也未再出現。有文獻報道,有些樣品在特定的色譜條件下可能存在結構的動態平衡而出現雙峰,這種雙峰是無法完全分離的,當改變色譜條件尤其是pH值時會使峰形正常[21]。因此,有必要充分考慮待分離樣品的酸度系數(pKa),以保證樣品在流動相pH條件下能以分子狀態存在[22]。由于奧卡西平的pKa為13.73[23],存在酰胺鍵,當流動相酸性較強時有可能出現上述文獻所述的動態結構平衡,故筆者調整水相pH后雙峰現象消失。
筆者在前期預試驗中發現,按“2.3.2①”項下方法制備供試品溶液進行重復性試驗時,奧卡西平含量的RSD值較大。進行含量測定時,樣品不能完全溶解,測得的含量較低,故本研究采用了不同的方法制備供試品溶液。
刻痕線的形狀、深度,刻痕片的形狀、大小、厚薄及曲率均可能影響片劑分劑量的準確性[1]。本研究結果顯示,仿制藥掰分時較費力且存在分劑量不均勻的現象,該結果與仿制藥半片制劑溶出均一性降低結果一致;仿制藥的脆碎度和分割質量損失均高于原研藥,表明仿制藥與原研藥的工藝參數可能存在差異[24],提示生產工藝可影響掰分后分劑量的準確性及溶出行為。
通過對不同企業奧卡西平原料藥進行比較后發現,國產原料藥與原研藥企業產原料藥雖然晶型相同,但晶體形貌有所不同。這不僅可能對粒子的表面積及粒子間的相互作用產生影響,還可能影響輔料混合以及制粒壓片,從而影響藥物的溶出度[25-26]。因此,對于難溶性藥物除需關注晶型[27]對藥物溶出度的影響外,還應當重視晶體形貌對溶出行為的影響。
綜上所述,奧卡西平刻痕片原研藥和仿制藥在溶出行為及均一性等指標上均存有一定差異,仿制藥的生產工藝和內在質量有待進一步提升。
參考文獻
[ 1 ] 國家藥品監督管理局藥品審評中心.仿制口服片劑功能性刻痕設計和研究的一般要求[EB/OL].(2019-07-04)
[2019-09-24]. http://www.cde.org.cn/news.do?method=
viewInfoCommon&id=314888.
[ 2 ] SHAH RB,COLLIER JS,SAYEED VA,et al. Tablet splitting of a narrow therapeutic index drug:a case with levothyroxine sodium[J]. AAPS PharmSciTech,2010,11(3):1359-1367.
[ 3 ] ROSENBERG JM,NATHAN JP,PLAKOGIANNIS F. Weight variability of pharmacist-dispensed split tablets[J].J Am Pharm Assoc:Wash,2002,42(2):200-205.
[ 4 ] FREEMAN MK,WHITE W,IRANIKHAH M. Tablet splitting:a review of weight and content uniformity part 1 of a 2-part series:next month:table splitting:a review of the clinical and economic outcomes and patient acceptance[J]. Consult Pharm,2012,27(5):341-352.
[ 5 ] 商國美,趙榮偉.我院住院處方最小規格劑量單位再分零使用情況分析[J].西北藥學雜志,2005,20(4):175-176.
[ 6 ] 吳婧倩,楊穎昕,呂志杰.我院住院藥房分劈藥片使用狀況調查及持續質量改進[J].實用藥物與臨床,2018,21(2):235-237.
[ 7 ] European Pharmacopoeia Commission. European pharmacopeia 9.0[S]. Strasbourg:European Directorate for the Quality of Medicine,2017:885.
[ 8 ] The United States Pharmacopeial Convention. The United States pharmacopeia 41:volume 2[S]. Baltimore:United Book Press,2018:3080、6457.
[ 9 ] FDA. Tablet scoring:nomenclature,labeling,and data for evaluation[EB/OL].(2013-03-13)[2019-06-04]. https://www.fda.gov/drugs/guidances-drugs/all-guidances-drugs.
[10] 金方方,尹婕,南楠.化學口服固體制劑仿制藥質量和療效一致性評價研究思考[J].藥物分析雜志,2018,38(4):575-581.
[11] 謝沐風.改善溶出度評價方法,提高固體藥物制劑水平[J].中國醫藥工業雜志,2005,36(7):447-451.
[12] 丁晶,汪昕.癲癇診療指南解讀[J].臨床內科雜志,2016,33(2):142-144.
[13] 胡利華,王曉玲.我院片劑分劑量使用現狀調查分析[J].兒科藥學雜志,2013,19(3):32-35.
[14] 國家藥典委員會.中華人民共和國藥典:二部[S]. 2015年版.北京:中國醫藥科技出版社,2015:1406.
[15] FDA. Drug database:dissolution methods[EB/OL]. (2004-
02-12)[2019-06-04]. https://www.accessdata.fda.gov/scripts/cder/dissolution/index.cfm.
[16] 國家食品藥品監督管理總局.普通口服固體制劑溶出度試驗技術指導原則[EB/OL].(2015-02-05)[2019-06-04].
http://www.sfda.gov.cn/WS01/CL0087/114286.html.
[17] 國家食品藥品監督管理總局.普通口服固體制劑溶出曲線測定與比較指導原則[EB/OL].(2016-03-18)[2019-06-
05].http://www.sfda.gov.cn/ws0l/cl0087/147583.html.
[18] 國家藥典委員會.中華人民共和國藥典:四部[S]. 2015年版.北京:中國醫藥科技出版社,2015:121.
[19] 張啟明,謝沐風,寧保明,等.采用多條溶出曲線評價口服固體制劑的內在質量[J].中國醫藥工業雜志,2009,40(12):946-955.
[20] 梁忠瑞.奧卡西平共晶的制備、表征與性質研究[D].北京:北京化工大學,2018.
[21] 龔時瓊.高效液相色譜分析中異常峰的分析與處理[J].實驗技術與管理,2010,27(6):37-42.
[22] 全紅娜,金松子,雷勇生,等.反相高效液相色譜中流動相選擇與優化的研究進展[J].現代藥物與臨床,2014,29(10):1190-1194.
[23] FDA. Approved drugs:oxcarbazepine[EB/OL].(2018-10-
13)[2019-06-04]. https://pdf.hres.ca/dpd_pm/00048247.
[24] 王芳,金曼,朱傳祥.影響片劑脆碎度的因素分析[J].齊魯藥事,2012,31(10):613-614.
[25] 申寶德,沈成英,徐玲霞,等.難溶性中藥納米混懸劑的體內外行為研究進展[J].中國中藥雜志,2018,43(19):3828-3833.
[26] 金瑞,程開生,李志云,等.纈沙坦原料顆粒形貌對纈沙坦氨氯地平片溶出的影響[J].中國粉體技術,2019,25(5):45-50.
[27] 譚菊英,黃麗麗,孫煜,等.吡羅昔康原料晶型對片劑溶出度的影響研究[J].藥物分析雜志,2017,37(3):550-557.
(收稿日期:2020-01-09 修回日期:2020-05-08)
(編輯:陳 宏)
