穩(wěn)定同位素標(biāo)記甲基毒死蜱-D6的合成研究
2020-06-18 07:41:54涂亞輝鄧曉軍鐘佳琪趙超敏徐仲杰
同位素 2020年3期
涂亞輝,鄧曉軍,鐘佳琪,趙超敏,羅 勇,潘 潔,徐仲杰
(1.上海化工研究院有限公司,上海 200062;2.國家同位素工程技術(shù)研究中心 上海分中心,上海 200062;3.上海出入境檢驗檢疫局 動植物與食品檢驗檢疫技術(shù)中心,上海 200135)
甲基毒死蜱又名雷丹,是一種有機磷殺蟲劑,在農(nóng)業(yè)生產(chǎn)上應(yīng)用較為廣泛,但同時會導(dǎo)致大氣、土壤、水等自然環(huán)境受到污染,并通過一定的富集作用,殘留至蔬菜水果等食品,引起人體產(chǎn)生一系列的中毒反應(yīng),導(dǎo)致有機磷農(nóng)藥殘留食品安全問題頻出。因此,世界上許多國家已經(jīng)針對不同食品類別制定了甲基毒死蜱的最大殘留檢測限量,使得這類化合物的殘留含量檢測引起廣泛關(guān)注[1-3]。
國內(nèi)外針對有機磷農(nóng)藥甲基毒死蜱殘留檢測分析方法主要有光電化學(xué)法、超高效液相色譜串聯(lián)質(zhì)譜法、氣相色譜串聯(lián)質(zhì)譜法、基質(zhì)固相分散法等[4-8],但是,這些分析方法存在一定的問題,如樣品的預(yù)處理復(fù)雜,基質(zhì)效應(yīng)影響較大等,嚴(yán)重影響了分析檢測結(jié)果的準(zhǔn)確性。因此,現(xiàn)代食品安全檢測需要開發(fā)更準(zhǔn)確有效的檢測方法。為了解決這些問題,采用同位素稀釋質(zhì)譜法(isotope dilution mass spectrometry, IDMS)可準(zhǔn)確有效地檢測食用品中有機磷農(nóng)藥甲基毒死蜱殘留含量[9-11]。本文旨在合成穩(wěn)定同位素標(biāo)記的甲基毒死蜱-D6作為檢測用內(nèi)標(biāo)試劑,廣泛地應(yīng)用于高效精確的農(nóng)藥殘留食品安全檢測,讓放心食品進(jìn)入生活,具有一定實用意義。
對于甲基毒死蜱的合成,有相關(guān)文獻(xiàn)[12-14]報道以O(shè),O-二甲基硫代磷酰氯為原料,與相應(yīng)的3,5,6-三氯吡啶-2-醇或其鈉鹽在水相體系或者兩相復(fù)合體系中,在相轉(zhuǎn)移催化劑的作用下發(fā)生取代反應(yīng),合成相應(yīng)的目標(biāo)產(chǎn)物。目前國內(nèi)外尚無關(guān)于穩(wěn)定同位素D標(biāo)記的甲基毒死蜱的合成及其作為穩(wěn)定同位素標(biāo)記內(nèi)標(biāo)試劑檢測甲基毒死蜱含量的相關(guān)報道。本研究結(jié)合O,O-二甲基硫代磷酰氯及甲基毒死蜱的工藝合成路線,以三氯硫磷和甲醇-D4為起始原料進(jìn)行親核取代反應(yīng),得到中間體O,O-二甲基硫代磷酰氯-D6,然后與3,5,6-三氯吡啶-2-醇作用得到目標(biāo)產(chǎn)物甲基毒死蜱-D6,可作為穩(wěn)定同位素內(nèi)標(biāo)試劑。
1 實驗部分
1.1 主要儀器與試劑
C-MAGHS7型加熱磁力攪拌器:德國IKA公司;RE-52A型真空旋轉(zhuǎn)蒸發(fā)器、DLSB-10/40型低溫冷卻循環(huán)泵:上海衛(wèi)凱儀器有限公司;600 MHz核磁共振譜儀:美國Bruker公司;GC 7890A氣相色譜儀:美國Agilent公司;Trace 1310-ISQLT頂空-氣相色譜-質(zhì)譜聯(lián)用儀:Thermo Fisher。
三氯硫磷:純度98%,上海化工研究院有限公司;乙醇、甲醇、丙酮、甲苯、二氯甲烷、乙醚、乙腈、苯、正己烷、乙酸乙酯:分析純(AR),國藥集團(tuán)化學(xué)試劑有限公司;氯化鈉、氫氧化鈉:AR,江蘇強盛化學(xué)試劑有限公司;1-甲基咪唑:純度98%,上海阿拉丁生化科技股份有限公司;硼酸、三乙基芐基氯化銨:98%,安耐吉化學(xué);甲醇-D4:豐度99atom%D,百靈威科技有限公司;3,5,6-三氯吡啶-2-醇:98%,上海特伯化學(xué)科技有限公司。
1.2 實驗方法
甲基毒死蜱-D6的總體合成路線示于圖1。
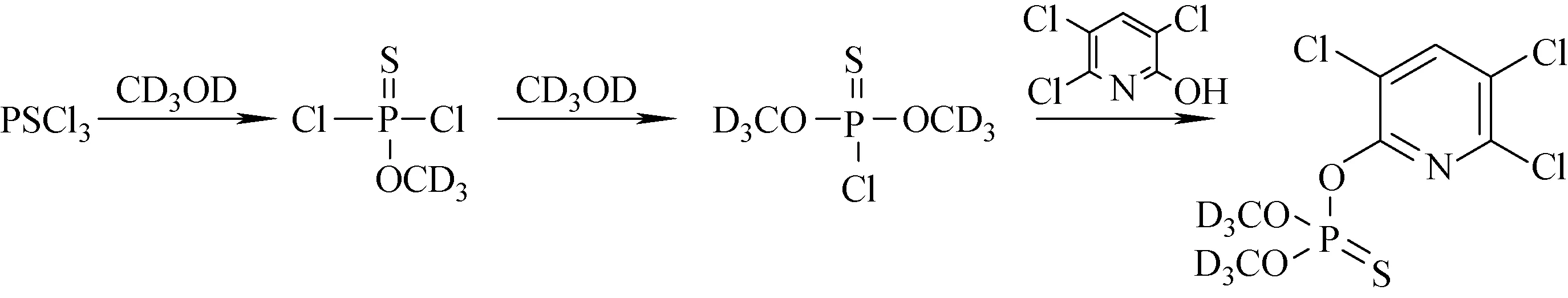
圖1 甲基毒死蜱-D6的總體合成路線
1.2.1O,O-二甲基硫代磷酰氯-D6(A3-D6)的合成 合成路線示于圖2。稱取6.67 g(39.0 mmol)三氯硫磷A1于25 mL的單口圓底燒瓶中,冰鹽浴冷卻至-5 ℃,在攪拌的條件下向反應(yīng)體系中緩慢滴加5.61 g甲醇-D4(156.0 mmol),保持溫度為-4~-6 ℃,攪拌15 min,然后向反應(yīng)體系中加入8 mL冰水以淬滅反應(yīng),保持溫度10 ℃以下,攪拌15 min,靜置分液,取有機相,得到無色液體4.9 g,即產(chǎn)物甲基二氯化物A2-D3,根據(jù)氣相檢測面積歸一法計算收率為27.0%(以甲醇-D4計),純度99.1%。直接投入下一步反應(yīng)。

圖2 O,O-二甲基硫代磷酰氯-D6(A3-D6)的合成路線
稱取4.90 g(28.9 mmol)的A2-D3,8 mL的丙酮于50 mL的三口燒瓶中,冰鹽浴冷卻至-5 ℃,在攪拌的條件下向反應(yīng)體系中緩慢地滴加4.70 g甲醇-D4(115.6 mmol),保持溫度為-4~-6 ℃,攪拌15 min,取10 g質(zhì)量分?jǐn)?shù)為30%的氫氧化鈉水溶液緩慢滴加至反應(yīng)體系中,同時檢測反應(yīng)體系中的pH,直至pH=10,停止滴加,取8 mL冰水加入到反應(yīng)體系淬滅反應(yīng),攪拌15 min后,靜置分液,取有機相,旋蒸除去丙酮試劑,得到無色液體3.1 g,即產(chǎn)物A3-D6,根據(jù)氣相檢測面積歸一法計算收率為23.6%(以甲醇-D4計)。
1.2.2甲基毒死蜱-D6的合成 合成路線示于圖3。稱取1.98 g(10.0 mmol)的3,5,6-三氯-2-吡啶醇,0.48 g(12.0 mmol)的氫氧化鈉于配有回流冷凝管和恒壓滴液漏斗的100 mL三口圓底燒瓶中,加入30 mL的水,加熱攪拌使得固體物質(zhì)完全溶解,降至室溫后向反應(yīng)體系中投入0.58 g(10.0 mmol)氯化鈉、0.12 g(2.0 mmol)硼酸、0.02 g(0.1 mmol)三乙基芐基氯化銨、8.2 mg(0.1 mmol)1-甲基咪唑和20 mL的二氯甲烷,通過滴液漏斗將1.83 g(11.0 mmol)O,O-二甲基硫代磷酰氯-D6滴加至反應(yīng)體系中,加熱至42 ℃,繼續(xù)反應(yīng)1.5 h,停止反應(yīng),冷卻至室溫,靜置分液,利用二氯甲烷進(jìn)行萃取(3×20 mL),用無水硫酸鈉干燥,過濾,旋蒸除去二氯甲烷,得到棕色固體。利用正己烷:乙酸乙酯=20∶1進(jìn)行柱層析提純,得到白色固體,再用無水乙醇重結(jié)晶,得到2.5 g白色晶體,氣相純度為98.3%,同位素豐度為99.0atom%D,收率為69.3%(以A3-D6計)。1H NMR(600 MHz, CDCl3)δ ppm 7.86(s, 1H)。
2 結(jié)果與討論
穩(wěn)定同位素標(biāo)記中間體O,O-二甲基硫代磷酰氯-D6的純度及收率是甲基毒死蜱-D6合成的關(guān)鍵因素,因此O,O-二甲基硫代磷酰氯的合成利用天然豐度原料進(jìn)行條件優(yōu)化[15-16]。
2.1 O,O-二甲基硫代磷酰氯合成的影響因素
2.1.1三氯硫磷和甲醇的摩爾數(shù)之比對甲基二氯化物合成的影響 三氯硫磷與甲醇的投料摩爾數(shù)之比對甲基二氯化物的合成有著重要的影響,見表1。

表1 三氯硫磷與甲醇的物質(zhì)的量比對反應(yīng)的影響
注: 1)n1∶n2=n(三氯硫磷)∶n(甲醇);2)收率為氣相檢測收率,以甲醇計。
由表1結(jié)果可知,甲醇與三氯硫磷在低溫條件下容易發(fā)生取代反應(yīng)生成甲基二氯化物即A2,目標(biāo)產(chǎn)物的含量隨著甲醇量的增加也逐漸提高,但甲醇過多,反而導(dǎo)致副產(chǎn)物A3含量增加,導(dǎo)致下一步甲基一氯化物A3(O,O-二甲基硫代磷酰氯)合成反應(yīng)的收率降低。因此,選擇三氯硫磷與甲醇的物質(zhì)的量之比為1∶4作為最佳優(yōu)化條件。
2.1.2甲基二氯化物和甲醇的物質(zhì)的量之比對甲基一氯化物合成的影響 甲基二氯化物(A2)和甲醇的物質(zhì)的量之比對甲基一氯化物(A3)合成的影響結(jié)果列于表2。
以甲基二氯化物(A2)為原料進(jìn)行反應(yīng),反應(yīng)活性降低,進(jìn)一步取代反應(yīng)生成甲基一氯化物較為困難,需加入NaOH水溶液調(diào)節(jié)溶液的酸堿性,促使取代反應(yīng)繼續(xù)進(jìn)行。隨著甲醇含量的增加,原料反應(yīng)完全,目標(biāo)產(chǎn)物A3含量也逐漸提高,但是相應(yīng)副產(chǎn)物的含量逐漸增多,影響目標(biāo)產(chǎn)物的收率。因此,選擇A2與甲醇的物質(zhì)的量之比為1∶4作為最佳優(yōu)化條件。

表2 甲基二氯化物(A2)和甲醇的物質(zhì)的量比對反應(yīng)的影響
注:1)n1∶n2=n(A2)∶n(甲醇);2)收率為氣相檢測收率,以甲醇計。
2.1.3反應(yīng)溶劑對甲基一氯化物(A3)合成的影響 甲基一氯化物的合成是雙分子的親核取代反應(yīng),甲氧基負(fù)離子在溶劑中易形成氫鍵被溶劑化,降低其反應(yīng)活性。向反應(yīng)體系中增加非質(zhì)子性溶劑可以避免被溶劑化,不僅可以降低甲醇用量,減少副產(chǎn)物三甲酯的產(chǎn)生,而且減少水解副反應(yīng)的發(fā)生[17]。
篩選不同的非質(zhì)子性溶劑,進(jìn)行條件優(yōu)化篩選,對甲基一氯化物合成的影響列于表3。

表3 非質(zhì)子性溶劑對A3合成的影響
注:收率為氣相檢測收率,以甲醇計。
由表3數(shù)據(jù)可以看出,在反應(yīng)體系中加入非質(zhì)子溶劑丙酮可以有效地提高甲基一氯化物(A3)的含量,利于甲基毒死蜱的合成。
2.1.4O,O-二甲基硫代磷酰氯A3合成穩(wěn)定性實驗 通過對O,O-二甲基硫代磷酰氯合成反應(yīng)條件的優(yōu)化篩選,第一步反應(yīng)選擇三氯硫磷與甲醇的物質(zhì)的量之比為1∶4,第二步反應(yīng)選擇A2與甲醇的物質(zhì)的量之比為1∶4及加入非質(zhì)子溶劑丙酮作為反應(yīng)最佳優(yōu)化條件,利用天然豐度甲醇進(jìn)行穩(wěn)定性重復(fù)實驗,具有較好的重復(fù)性,結(jié)果列于表4。
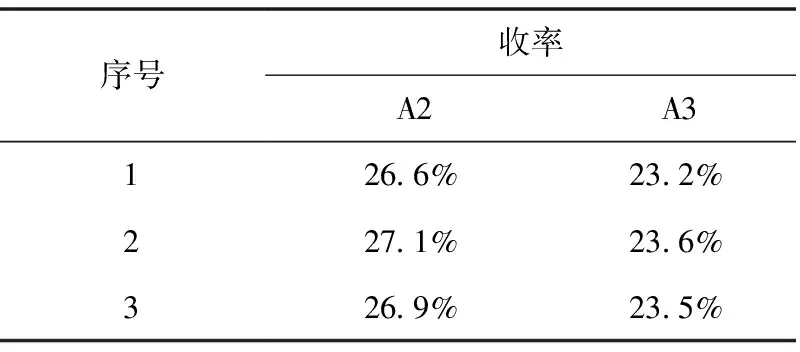
表4 穩(wěn)定性重復(fù)實驗數(shù)據(jù)
注:收率為氣相檢測收率,以甲醇計。
2.2 甲基毒死蜱的合成
根據(jù)甲基毒死蜱的合成工藝[13],按照n(A3)∶n(3,5,6-三氯-2-吡啶醇)∶n(氫氧化鈉)∶n(氯化鈉)∶n(硼酸)∶n(三乙基芐基氯化銨)∶n(1-甲基咪唑)=1.00∶1.10∶1.20∶1.00∶0.20∶0.01∶0.01的物質(zhì)的量比,二氯甲烷和水作為溶劑,回流1.5 h,利用二氯甲烷進(jìn)行萃取,進(jìn)行柱層析得到目標(biāo)產(chǎn)物,收率為70.1%(以A3計),純度為98.3%。
2.3 豐度實驗
經(jīng)過多次實驗條件優(yōu)化,確定最佳反應(yīng)合成路線,經(jīng)重復(fù)實驗驗證后,以甲醇-D4為同位素起始原料進(jìn)行豐度實驗合成甲基毒死蜱-D6,以消耗的甲醇-D4計,甲基毒死蜱-D6的總收率為44.2%,純度為98.3%,同位素豐度為99.0atom%D。
2.4 產(chǎn)品結(jié)構(gòu)表征
甲基毒死蜱-D6的1H NMR譜圖如圖4所示。1H NMR(600 MHz, CDCl3)δ ppm 7.86(s, 1H)。譜圖積分為1個氫,與天然豐度的甲基毒死蜱進(jìn)行對比,甲氧基上的氫被氘原子取代,與甲基毒死蜱-D6結(jié)構(gòu)一致。
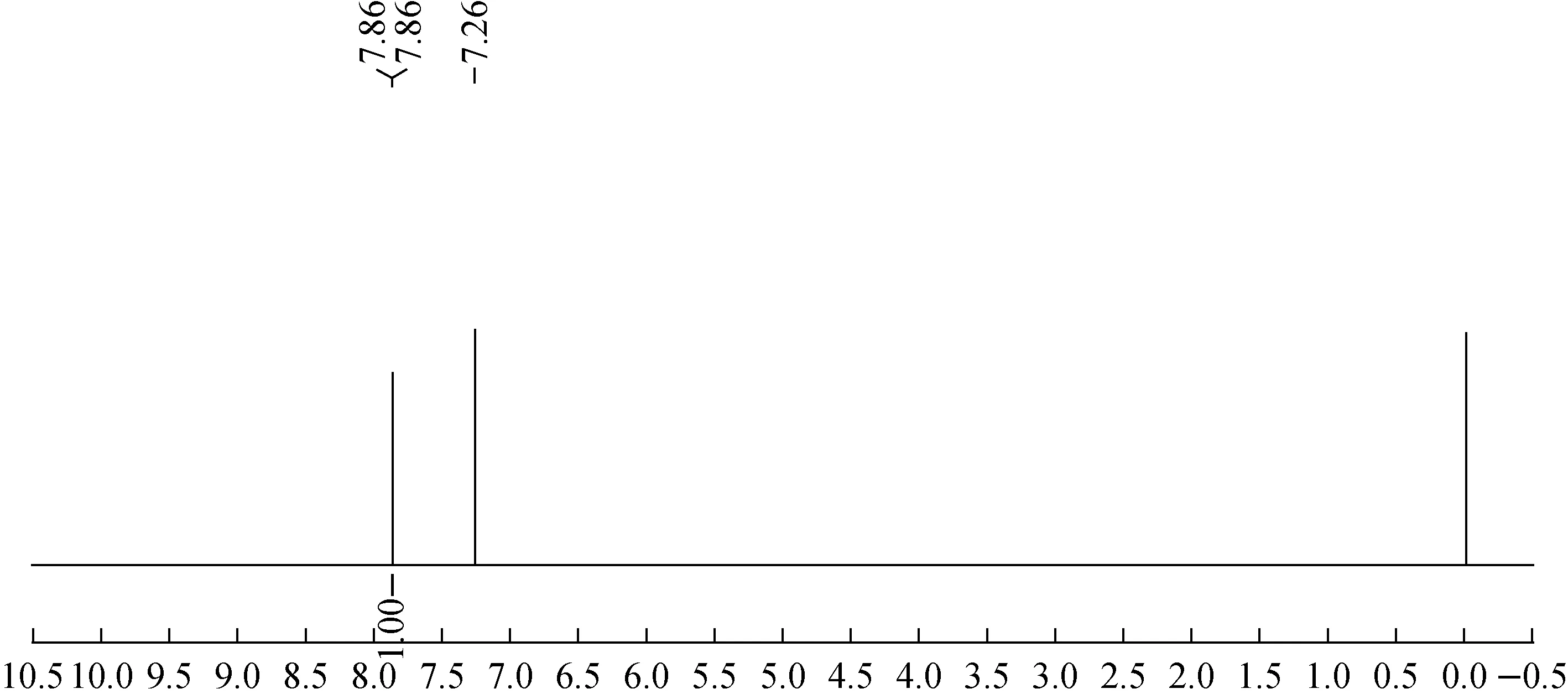
圖4 甲基毒死蜱-D6的1H NMR譜圖
通過對比產(chǎn)品與天然豐度甲基毒死蜱的MS數(shù)據(jù),甲基毒死蜱-D6產(chǎn)品的GC-MS圖譜(圖5)中主峰m/z=131.0,為甲基毒死蜱-D6減去3,5,6-三氯-2-吡啶醇,即[M-198.4+1]+峰,其質(zhì)量數(shù)131.0≈328.47-198.4+1。其中,328.47為甲基毒死蜱-D6的相對分子質(zhì)量,198.4為3,5,6-三氯-2-吡啶醇的相對分子質(zhì)量,0.07的質(zhì)量數(shù)在儀器誤差之間。可以確定合成的產(chǎn)物為甲基毒死蜱-D6。以甲基毒死蜱-D6的[M-198.43+1]+峰強度計算,豐度為99.0atom%D。
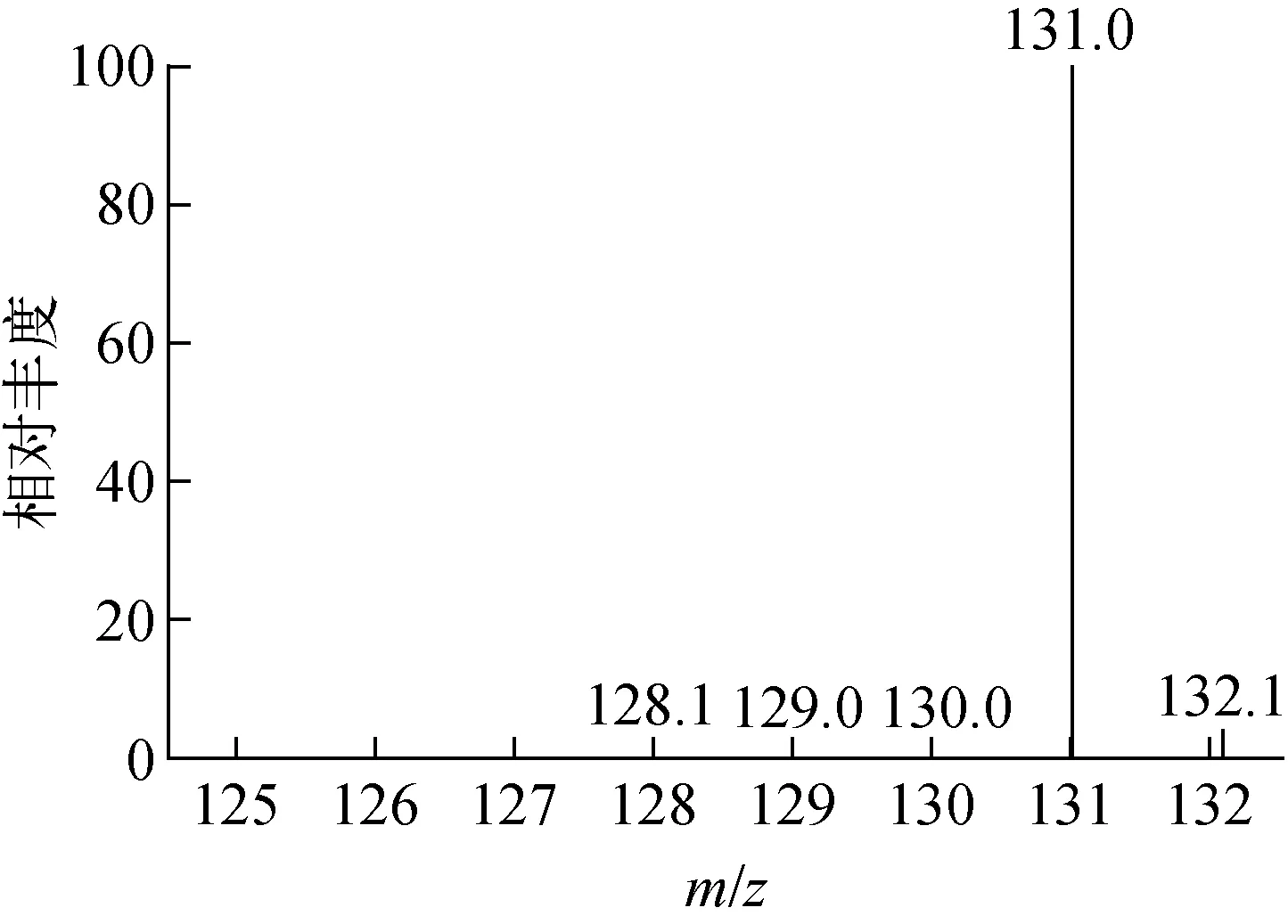
圖5 甲基毒死蜱-D6的GC-MS譜圖
3 結(jié)論
本文以甲醇-D4為穩(wěn)定同位素標(biāo)記原料,得到穩(wěn)定同位素標(biāo)記中間體O,O-二甲基硫代磷酰氯-D6。經(jīng)兩相復(fù)合體系發(fā)生取代反應(yīng)得到甲基毒死蜱-D6,利用簡單易得的甲醇-D4作為原料,以三步的取代反應(yīng),合成穩(wěn)定同位素標(biāo)記的甲基毒死蜱-D6氘代試劑,目前無相關(guān)的文獻(xiàn)報道。以消耗的甲醇-D4計,甲基毒死蜱-D6收率為44.2%,純度為98.3%,同位素豐度為99.0atom%D,該合成工藝簡單易操作,適用于實驗室合成,氘代產(chǎn)品可作為內(nèi)標(biāo)標(biāo)準(zhǔn)試劑應(yīng)用于食品安全農(nóng)藥殘留檢測。
