土壤中砷的來源及遷移釋放影響因素研究進展①
2020-06-15 01:47:24安禮航劉敏超張建強陳志良
土壤 2020年2期
關鍵詞:研究
安禮航,劉敏超,張建強,黃 玲,陳志良*
(1 五邑大學生物科技與大健康學院,廣東江門 529020;2 生態環境部華南環境科學研究所,廣州 510655;3 廣東省農田重金屬污染土壤治理與修復工程技術研究中心,廣州 510655)
砷是具有急性和慢性毒性有毒準金屬元素,被國際癌癥研究機構(IARC)定義為I 類致癌物質。人體砷中毒會導致肺損傷、外周神經損傷、皮膚病或心血管病等癥狀,嚴重則致癱瘓和致癌致畸[1]。土壤中的砷含量和飲用水中的砷濃度具有密切聯系,1993 年世界衛生組織(WHO)建議將飲用水中砷含量標準由50 μg/L降低為10 μg/L,中國、印度以及美國等國家部分水源中砷含量均超過該標準[2]。全世界約有2 億人的飲用水安全受到高砷暴露的威脅[3],尤其是東南亞國家,中國約有1 958 萬人生活在地下水砷超標的高風險地區。
我國砷污染問題尤為突出,地下水含砷量在10 μg/L以上的地區總面積達5.8×105km2,土壤中砷平均含量為11.2 mg/kg,約為世界平均值(7.2 mg/kg)的1.5倍[4]。土壤中砷來源于自然本底與人類活動,特別是由于人類活動,如礦山開采、冶煉、施肥、農藥等導致大量的砷進入土壤環境。2014 年全國土壤污染狀況調查公報顯示,全國土壤總的超標率為16.1%,鎘、汞、砷、銅、鉛5 種無機污染物點位超標率分別為7.0%、1.6%、2.7%、2.1%、1.5%,砷為全國土壤重金屬污染物中的首要污染物[4]。水稻是我國受砷污染的主要糧食作物,我國人均砷攝取量約為42 μg/d,遠高于西方國家,相當于美國、加拿大、澳大利亞及法國總砷攝入量的4 倍 ~ 4.7 倍,其中通過大米攝入的砷占總砷攝入量的60%[5],較高含砷量的稻米使得以之為主食的人民的健康受到嚴重威脅。近年來許多學者致力于砷的地球化學行為和降低砷對人體毒害的研究,已取得了一系列的成果,但砷的環境行為受諸多因素的影響,因素間的相互作用機理復雜,目前對這方面鮮有綜合性分析。本文綜述了環境中砷的來源及影響砷遷移轉化的主要因素,為解決土壤砷污染問題提供依據。
1 砷的主要來源
1.1 人為來源
1.1.1 工業采礦和排廢 礦物資源開發和工業廢物排放(如冶煉和化石燃料燃燒產物)是土壤砷的主要人為來源。常見的礦物資源有含砷黃鐵礦增生體(Fe(S,As)2)、毒砂(FeAsS)、斜方砷鐵礦(FeAs2)、雄黃(AsS)、雌黃(As2S3),輝砷鈷礦(CoAsS)、紅砷鎳礦(NiAs)和臭蔥石(FeAsO4·2H2O)[6],其中含砷量相對較高的是含砷黃鐵礦增生體,其砷含量超過 100 g/kg(最高達190 g/kg)[7],含砷量最高的是硫化物和氧化物礦物(鐵礦)(表1),其他常見的硫酸鹽成巖礦物(碳酸鹽和硅酸鹽)、硅酸鹽礦物(包括石英、長石、云母、閃石)和碳酸鹽礦物中的砷含量往往較低(一般小于5 mg/kg)。有色金屬冶煉過程排廢是最重要的砷污染源之一,全球該行業每年排放砷近10 萬t,其中90% 左右的砷來自銅、鉛、鋅等有色金屬冶煉行業(圖1)[9]。
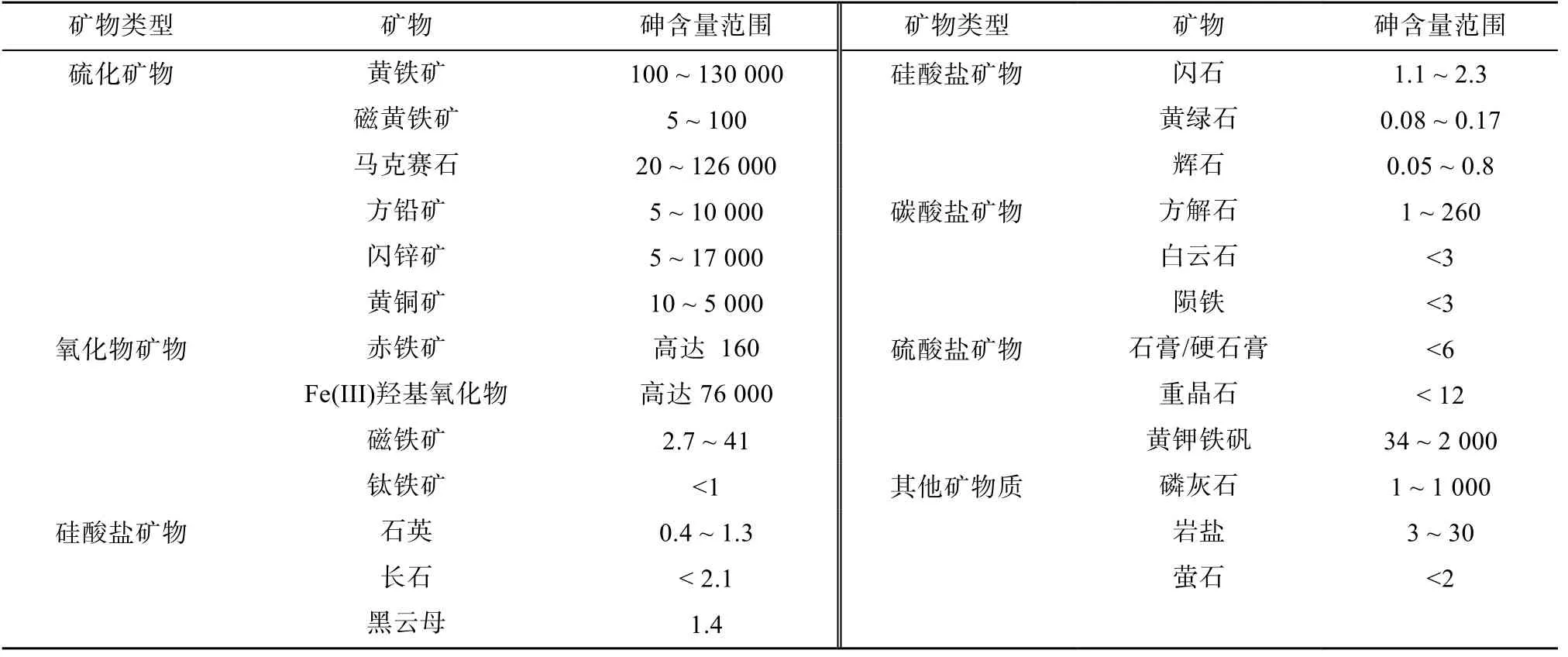
表1 礦物中砷的典型含量范圍[6-8] (mg/kg)Table 1 Typical ranges of As concentrations in minerals
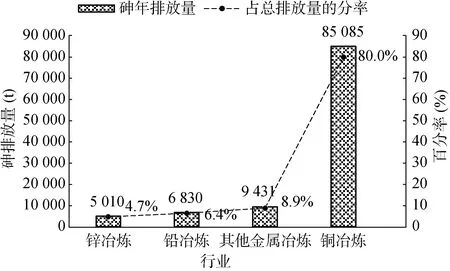
圖1 全球冶金工業砷年排放量Fig. 1 As emissions from the global metallurgical industry
1.1.2 農業排放 農業生產過程中使用農藥和磷肥是土壤砷的另一個重要來源。牲畜養殖中常用多種含砷化合物如洛克沙胂等作為飼料添加劑,該類添加劑經動物代謝后產生含砷化合物及糞便經農用釋放進入土壤[10]。歷史上將砷類農藥施用于水果作物后,在果園土壤中檢測砷含量高達732 mg/kg。長期使用磷肥也會使土壤中富集砷。受工業污染的沉積物和土壤,其砷含量比自然條件下沉積物和土壤的本底值高幾個數量級,達數千甚至上萬mg/kg (表2)。
1.2 自然來源
含砷硫化物巖石和氧化物巖石經風化和雨水沖蝕等過程釋放可溶態砷是土壤砷的主要自然來源。土壤和沉積物是最重要的砷匯,地殼中平均豐度約為1.5 mg/kg,土壤中砷含量約為5 ~ 15 mg/kg,全球土壤平均為7.2 mg/kg[8]。根據砷含量范圍可將土壤沉積物和巖石分為3 類,第一類在5 mg/kg 以下,包括火成巖、變質巖、沉積巖中的砂巖以及河床沉積物等;第二類在5 ~ 15 mg/kg 范圍,包括典型泥質巖(板巖、千枚巖)、沉積巖、泥質沉積物、河流沉積物、海洋黏土、煤礦、泥炭和黏土等,較高的含量反映了這類物質中存在一定比例的硫化物礦物、氧化物和有機物;第三類砷含量最高(20 mg/kg 以上),一般為硫鐵氧化物,包括頁巖、磷灰巖(達400 mg/kg)、鐵質地層和富鐵沉積物等(表3)。
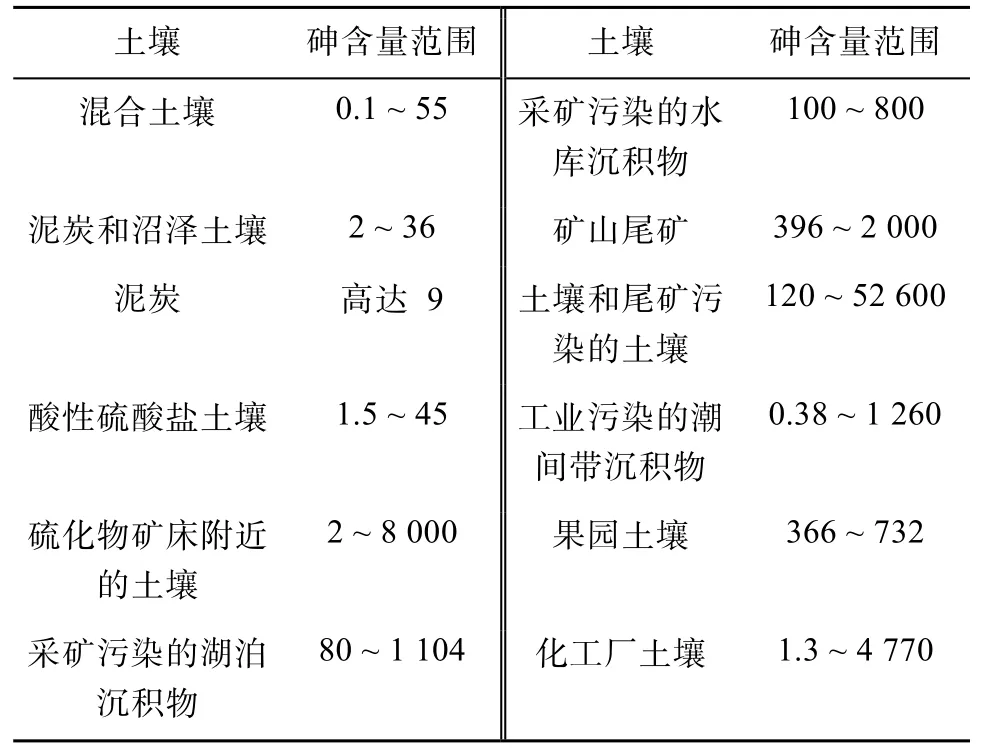
表2 土壤中砷的典型含量范圍[8] (mg/kg)Table 2 Typical ranges of As concentrations in soils
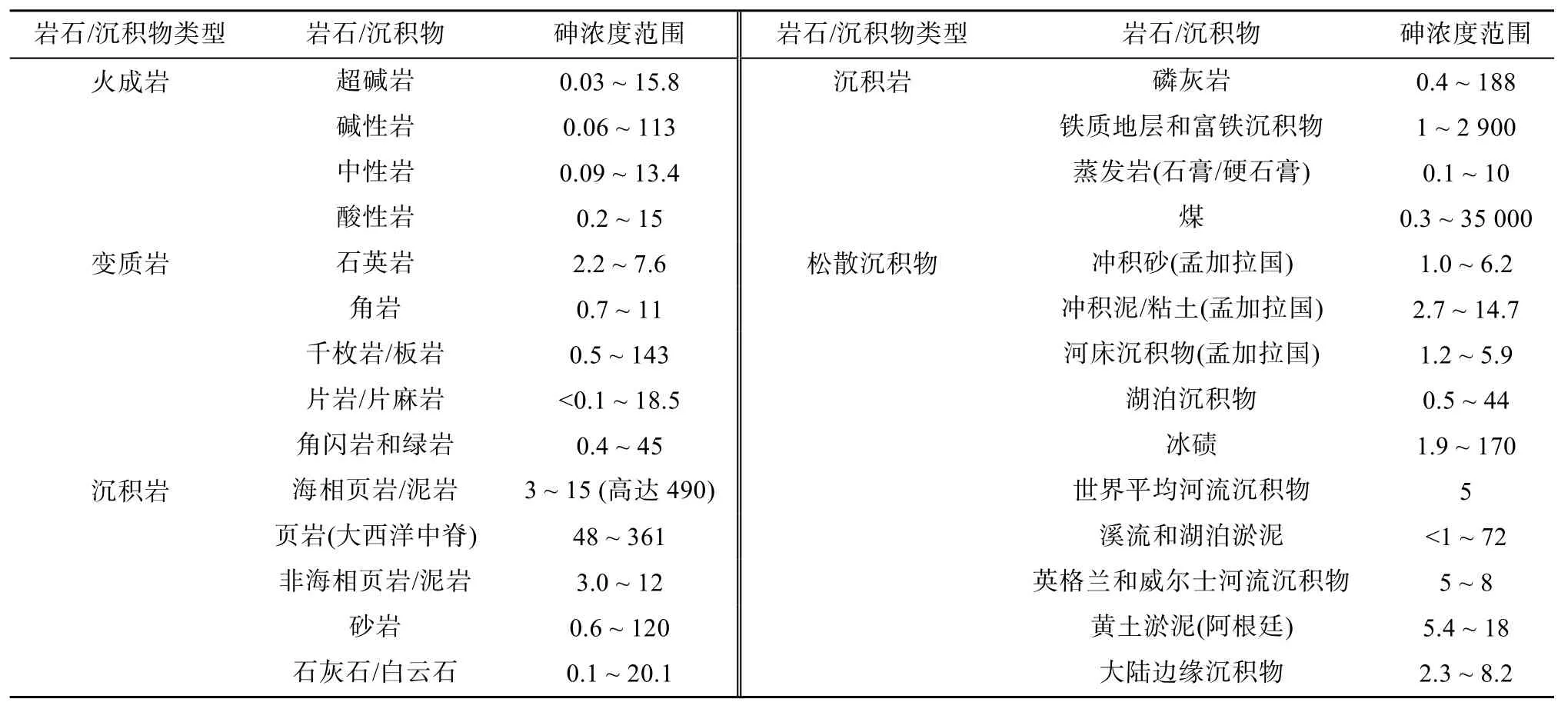
表3 巖石、沉積物中砷的典型含量范圍[8, 11] (mg/kg)Table 3 Typical ranges of As concentrations in rocks, sediments
綜上,土壤中砷的來源主要是人類工農業活動和自然巖石風化,自然來源相對貢獻較小,相比之下,工業污染導致土壤中砷含量高出幾個數量級。需指出的是,土壤中砷的含量水平、分布特征、砷的地球化學特性也在很大程度上與其所處的環境條件密切相關。
2 影響砷遷移的因素
2.1 金屬礦物對砷遷移的影響
2.1.1 鐵礦物對砷的影響 在自然體系中,鐵礦物的類型和分布、還原條件的持續時間[12]以及鐵礦物還原轉化的速率和程度是決定砷固定或釋放的主要因素。Fe(III)(羥基)氧化物還原釋放砷被確定為導致土壤和沉積物中孔隙水砷濃度升高的罪魁禍首,已有大量文獻報道了其相關性,如Huang 等[13]發現總鐵與總砷具有顯著的相關性(r=0.70,P<0.05);Bennett等[14]研究表明土壤溶液中Fe(II) 濃度和As(III) 濃度呈極顯著正相關(r=0.896,P<0.001);進一步研究指出Fe(II)和總砷之間有顯著的正相關性(r=0. 868,P<0.001),二者存在耦合釋放的關系[15];Honma 等[16]研究也證實土壤溶液中Fe(II) 與總砷濃度存在二次函數關系:[As]= -0.002 4[Fe(II)]2+ 0.312 5[Fe(II)] +3.588 6。由此可見,鐵礦物的還原性溶解和砷的遷移轉化具有緊密的聯系。
鐵砷循環耦合關系及其機理主要包括兩個方面,即鐵砷耦合的氧化還原過程和鐵礦物對砷的吸附解吸過程。1)鐵砷耦合的氧化還原過程(圖2)。鐵砷耦合的氧化還原過程包括由鐵還原菌的作用引起鐵氧化物的還原溶解導致砷的釋放和吸附在鐵氧化物上砷的直接還原[17]。相比于土壤溶液中砷的還原,吸附在鐵礦物表面上砷的直接還原半衰期極高,因此大量研究集中在鐵礦物還原驅動砷釋放。微生物介導的鐵還原過程包括同化鐵還原和異化鐵還原過程,同化鐵還原指Fe(III) 被運輸到細胞體內被鐵還原酶所還原;異化鐵還原是指微生物將電子轉移至細胞外的鐵氧化物表面,將Fe(III) 還原成Fe(II) 的過程[18],其中異化鐵還原是主要過程。在厭氧條件下,異化鐵還原耦合砷氧化,Fe(III) 還原成Fe(II) 溶解,其反應式為CH3COO–+2Fe2O3+4Fe(OH)3+ 15H+→8Fe2++2HCO–3+14H2O;同時吸附的As(V) 進行溶解,溶液中As(V)濃度先增加,后在異化砷的作用下還原成As(III),其反應式為CH3COO–+2HAsO2–4+2H2AsO–4+5H+→4H3AsO3+2HCO–3[19]。異化砷是指氧化乙酸、有機質等電子供體耦合As(V) 還原,同時為微生物提供能量[20]。由于Fe(III) 和As(V) 同為電子受體,能替代鐵還原菌或砷還原菌,因此二者之間存在競爭還原關系,在足夠電子供體情況下鐵砷先后被還原,但在電子供體不足的情況下,鐵砷之間的還原就存在競爭(圖3)。有機質能提高土壤溶液中砷的有效性[21],還能介導氧化還原電位的變化以順利促使砷的活化[14]。而有機質耦合鐵或砷的還原中,二者哪個更有耦合有機質降解的優勢有待進一步研究。
2)鐵礦物對砷的吸附解吸過程。在鐵還原過程中發生3 個競爭過程,共同決定了As(III) 的溶解濃度:①孔隙水置換(流動)過程中化學失衡促進As(III) 解吸;②鐵相的溶解釋放As(III);③次生鐵相固定As(III)[12]。故在砷解吸過程中也通過吸附、(共)沉淀或兩個反應同時進行使砷重新固定[22]。先前存在大量報道鐵(氫)氧化物還原溶解導致砷解吸釋放的研究[14-16],但最近研究發現與之相反的情況,即隨著水鐵礦的還原轉化,其重結晶為更穩定的次生Fe(II) 礦物相,增加了As(III) 和As(V) 的吸附固定[23],且進一步的研究表明水鐵礦還原轉化初始階段增強As(III)固定(相對于非生物系統),而長時間的還原則導致砷的解吸[12]。這說明鐵氧化物的還原溶解不一定會導致砷的遷移,Fan 等[24]也發現當As(V) 引入缺氧土壤時,能與Fe(II)/ Fe(III)快速共沉淀(形成砷酸鐵和砷酸亞鐵)阻止其還原為As(III),該過程是控制砷固定的主要機制。然而當砷對次生Fe(II)/ Fe(III) 礦物的結合能力較低時,吸附砷的Fe(III) 礦物的微生物還原則會導致砷的遷移。例如,在含As(III) 的Fe(III)礦物的微生物還原中,發現As(III) 強烈的遷移;但在含As(V) 的Fe(III) 礦物的微生物還原中則導致砷的固定,這種差異可歸因于As(III) 和As(V) 與次生Fe(II)/ Fe(III) 礦物的結合能力的差異[25]。此外,鐵還原菌FeRB 的活動可促進上述過程,一方面使含砷鐵礦物和水稻根表鐵膜[26]還原溶解而促進砷的釋放,另一方面在鐵還原過程中誘導形成對砷具有高結合能力的結晶次生鐵礦物而促進砷的固定[25]。這些研究說明了鐵氧化物的還原溶解對砷釋放或固定的凈效應取決于砷對次生鐵礦物的結合能力。
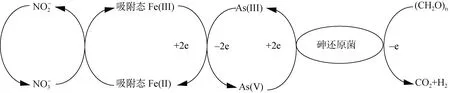
圖2 微生物鐵還原耦合砷氧化過程Fig. 2 Microbial Fe reduction coupled As oxidation process
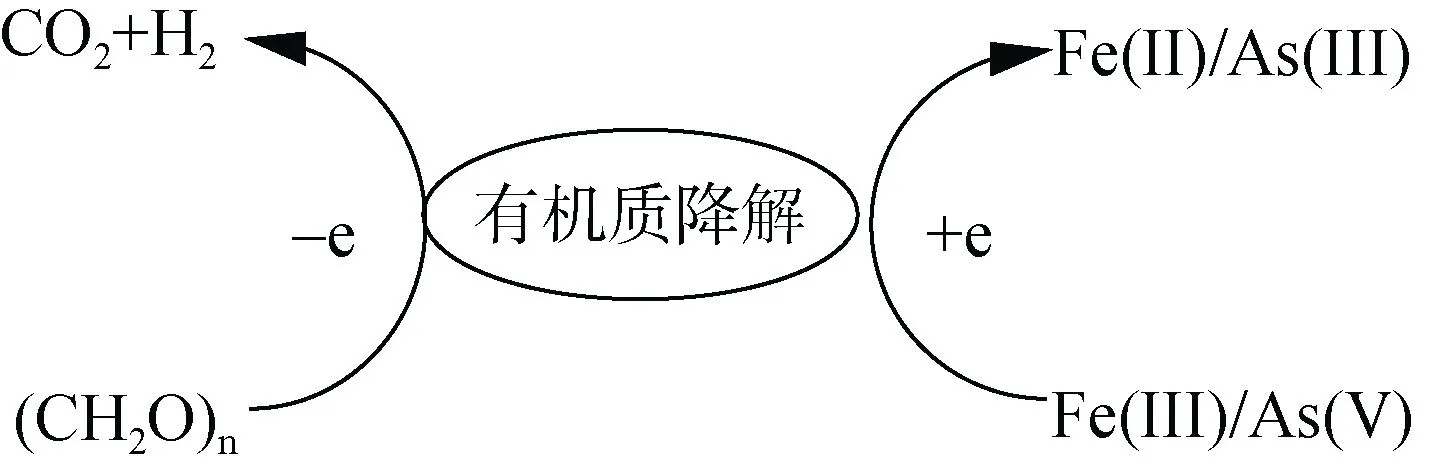
圖3 鐵砷競爭耦合還原Fig. 3 Fe-As competitive coupling reduction
沉積物和土壤環境中的鐵礦物存在廣泛的吸附砷過程[27],鐵礦物吸持砷是一種有效的自然衰減過程,可顯著降低高砷水體(如酸性礦山廢水)中的砷濃度。例如,大多數土壤和沉積物中的鐵礦物能與As(V)強烈結合,尤其是將其吸附到具有高比表面積的鐵礦物上[28],如纖鐵礦(β-FeO(OH)/γ-FeO(OH))和水鐵礦(Fe5HO8·4H2O)(條件分別為2.5≤pH≤6 和6≤pH≤8)[29];而As(III) 的吸附更具特異性,其與Fe(III)(氫)氧化物結合最明顯[30],并且在近中性和堿性的靜水(間歇)條件下比As(V) 更廣泛地吸附在鐵(氫)氧化物和磁鐵礦上[23,31]。不同類型的鐵礦物對砷的吸附機制以及特點見表4。
2.1.2 鋁氧化物對砷的影響 鋁氧化物對砷的固定起著重要作用,其作用受到氫氧化鐵強烈的干擾,因此研究通常將鋁和鐵綜合一起討論。Goldberg 和Johnston[41]研究了As(III)、As(V) 與鐵、鋁(氫)氧化物的相互作用并指出吸附機理是:As(V) 在鋁(氫)氧化物或鐵(氫)氧化物表面形成親和力較強的內層吸附;而As(III)在鐵(氫)氧化物表面同時形成內層和外層吸附,但在鋁(氫)氧化物表面只能形成外層吸附,因此導致了As(III)極易從鋁(氫)氧化物表面解吸。鋁和鐵對砷的吸附和釋放影響機制如圖4 所示。

表4 土壤鐵礦物類型對于砷的吸附機制以及特點Table 4 Adsorption mechanisms and characteristics of As on soil Fe mineral types
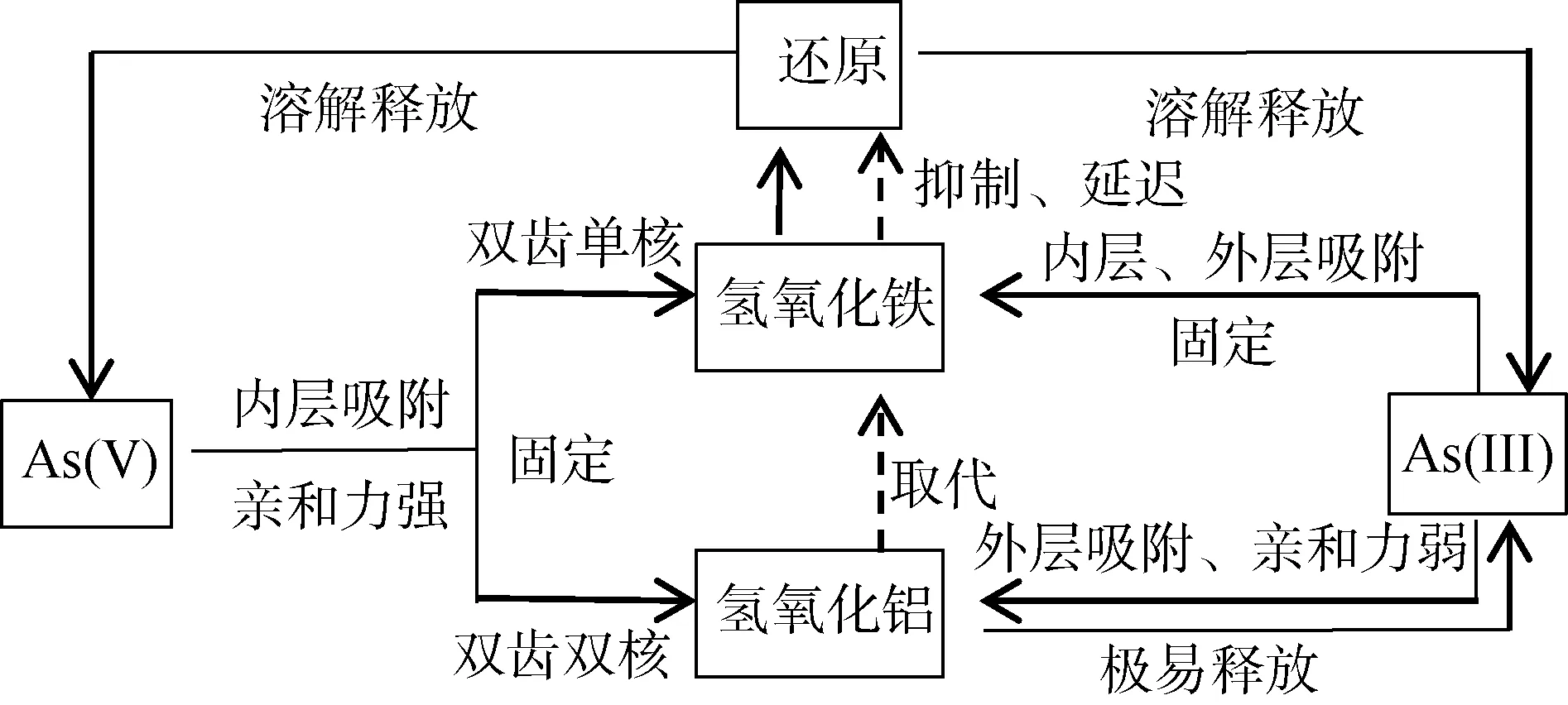
圖4 鋁鐵氫氧化物對砷的吸附及影響機制Fig. 4 Mechanism of adsorption and influence of Al and Fe hydroxide on As
在pH<7.5、同等摩爾比的條件下,鋁(氫)氧化物和鐵(氫)氧化物對As(V) 的吸附能力大致相同;鋁(氫)氧化物極易吸附As(V),吸附最大值在pH 4 ~4.5,而幾乎不吸附As(III)[42],因此在任何pH 條件下,鐵(氫)氧化物對As(III) 的吸附能力更強[43]。研究表明使用0∶1 或1∶4 的鋁氫氧化物∶鐵氫氧化物可以實現同等效果的最大As(V) 吸附率,而當鋁鐵摩爾比大于1∶4 時,隨著其摩爾比的增加,形成結晶鋁(氫)氧化物(三羥鋁石和三水鋁石)導致As(V) 和As(III) 的吸附量減少,尤其As(III) 的解吸增加[44]。因鋁(氫)氧化物對As(III) 的吸附幾乎可以忽略不計,故0∶1 的鋁∶鐵氫氧化物比1∶4 的鋁∶鐵氫氧化物吸附As(III) 效果更好[44]。進一步研究指出,鐵(氫)氧化物介質中As(V) 還原形成的As(III) 可再次被鐵(氫)氧化物吸附,向水相中的釋放量非常低,但當鋁引入后,As(III) 向環境的釋放明顯增加,尤其在鋁(氫)氧化物體系中,幾乎全部的As(III) 均被釋放[45]。因此,鐵(氫)氧化物比鋁(氫)氧化物及兩者混合物吸附砷的能力更強。
另一方面,As(V) 對于鋁(氫)氧化物的鍵合力比鐵(氫)氧化物更強。砷鍵合在結晶鋁(氫)氧化物上是雙齒雙核,而砷對無定形羥基鐵(氫)氧化物的鍵合是雙齒單核[46];研究發現含鋁/砷的土壤與砷鋁石(AlAsVO4·2H2O)的EXAFS 光譜非常相似,其中砷以As(V) 形式存在,第一個As—O 配位殼體在1.70? ±0.01? 處填充4 個氧鄰位,砷在3.18? ± 0.02? 處有2個鋁鄰位,這與雙齒雙核的As—O—Al 鍵(AsO4單元的兩個O 原子分別鍵合到兩個Al 原子上)一致[46]。此外,由于鐵存在氧化還原過程[47],鐵(氫)氧化物及其吸附的砷易溶解釋放。土壤中大部分鐵(氫)氧化物都被鋁取代[44],共沉淀的鋁/鐵氫氧化物可能對于砷的吸持更有利,因為結構中存在鋁取代的鐵(氫)氧化物的還原被延遲,降低溶解速率[45]。如一項比較鋁取代的針鐵礦和純針鐵礦的細菌還原溶解研究證實,當在針鐵礦中存在鋁取代時,溶解速率更慢[48]。由此可見,在還原環境中,鋁/鐵氫氧化物混合物對于吸附砷可能比鐵氫氧化物更有利。
2.1.3 錳氧化物對砷的影響 錳氧化物是一種非生物強氧化劑,已被證明可以氧化As(III)、Fe(II)、Co(II)、U(IV)、Cr(III) 和有機物質[49]。富錳的地層通常形成于地下水位頻繁波動的區域,例如淺海區和淺層含水層[50]。沖積平原和泛濫平原中富錳物質的沉積過程也會產生富錳環境[51]。自然中重要的錳化物包括水鈉錳礦物([Na,Ca,Mn(II)]Mn7O14·2.8H2O)、鋰輝石(LiAl2[Mn(IV)2Mn(III)]O6(OH)6)(雙層結構)、鋇鎂錳礦((Ca,Na,K)0.3~0.5[Mn(IV),Mn(III),Mg]6O12·3~4.5H2O)(隧道結構)[52],其零電荷低點(pH 1.7 ~ 3.5)使其成為各種重金屬(Zn、Ni、Co 和Pb)的有效吸附劑[53]。錳氧化物的還原在熱力學上比Fe(III) 和As(V) 更有優勢,因其具有相對較高的氧化還原電位(約400 mV)[54]。錳化物具有與鐵相似的氧化還原化學性質,在氧化條件下能迅速氧化 As(III) 和Fe(II),在土壤和沉積物的含氧/缺氧循環過程中與砷的固定/遷移有很強的聯系[55]。錳氧化物對砷的影響過程如圖5 所示。
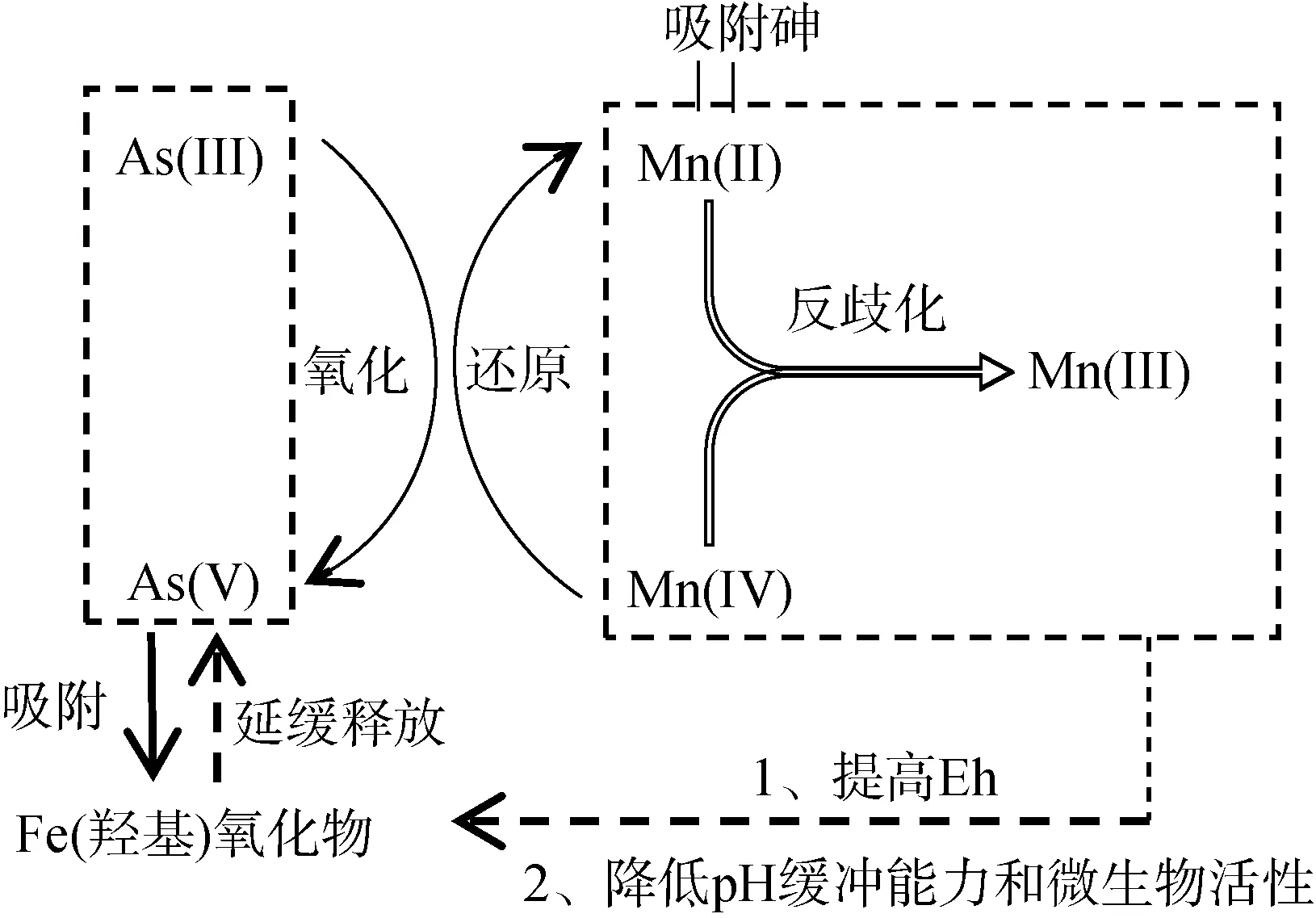
圖5 錳氧化物對砷的影響過程Fig. 5 Effect of Mn oxide on As
錳氧化物將As(III) 氧化成As(V) 涉及多個同時反應,包括Mn(IV) 還原為Mn(II)、Mn(II) 吸附以及Mn(IV) 和Mn(II) 反歧化為Mn(III) 反應[56]。除了直接氧化As(III) 的作用外,錳氧化物還可以通過將氧化還原電位提高到更高的水平來延緩含砷的鐵(羥基)氧化物的還原溶解以及砷向孔隙水的釋放[57]。例如,在一個砷污染洪泛區土壤的研究中,Ehlert等[58]發現,水鈉錳礦添加劑可促進Fe(II) 氧化形成新沉淀的Fe(III)氫氧化物,其作為As(III) 的高效吸附劑進一步延遲了砷和鐵的還原,從而顯著降低砷浸出率;Xu 等[59]也觀察到了相同的結果,錳氧化物含量較高的土壤表現出較少的砷遷移特性,原因是錳氧化物能夠減緩土壤淹水時氧化還原電位的下降,并使固相和溶液相中保持較高的砷酸鹽/亞砷酸鹽比率從而降低砷的遷移。此外,高濃度錳氧化物還可降低土壤還原期間的pH 緩沖能力,使微生物活性下降[58]。由此可見,富集氧化錳的土壤層能夠有效降低砷的遷移。
但是,在微生物還原條件下錳氧化物對As(III)的氧化是有限的[56-57],微生物作用將As(V) 還原成As(III),由于錳的表面鈍化反應,As(III) 不再被水鈉錳礦或Mn(III) 的轉化產物所氧化。盡管也有研究發現亞錳酸鹽也可氧化As(III)[60],但是貢獻不大。表面鈍化反應可能是由于水鈉錳礦轉化為亞錳酸鹽的緩慢動力學導致Mn(II) 和Fe(II) 吸附在水鈉錳礦表面,分別導致還原歸中反應(Mn(IV) 和Mn(II) 反歧化為Mn(III))和表面沉淀[58];另一原因是微生物代謝產物積累覆蓋了錳氧化物活性表面。因此錳氧化物對As(III) 的氧化能力在淹水土壤和沉積物中可能受到限制。在這種環境下,吸持在固相中的As(III) 的穩定性是短暫的,進一步的土壤還原將導致次生Fe(III) 相的異化還原,最終導致As(III) 和Fe(II) 的釋放。但是,在氧化還原條件頻繁變化的情況下,富含錳氧化物的土壤可能成為長期的砷匯,使砷浸出風險降低,促進富錳氧化物的土壤層中砷的富集。因此,錳氧化物是調控污染土壤中砷浸出風險的重要因素之一,在治理砷污染方面很有前景。
2.2 陰離子對砷遷移的影響
2.2.1 硫循環對砷的影響 砷可形成不溶性硫化物礦物,包括雌黃(As2S3)、雄黃(AsS)和毒砂(FeAsS),也可形成多種金屬硫化物,包括四方硫鐵礦(FeS)、膠黃鐵礦(Fe3S4)和黃鐵礦(FeS2),并可在這些硫化物上作為吸附絡合物存在[61]。硫化物礦物的形成通常通過提供電子供體以及在某些情況下向沉積物和土壤中添加無機硫酸鹽來刺激土壤中硫酸鹽還原菌(SRB)的活性來實現。通過該過程,溶解態的砷可以形成含砷硫化物固體[61]。微生物硫酸鹽還原一般不直接影響溶解態砷濃度[62],而硫酸鹽化學還原形成的硫化物可以通過共沉淀與吸附共同作用降低砷濃度[63]。另一方面,地下水中常見的硫氧化劑包括溶解氧、硝酸鹽和Fe(III)(低pH 值時)可以使含砷硫化物再次氧化[64],促進砷釋放到地下水中。根據已發表的關于硫酸鹽還原的研究,提出一個描述硫循環過程的概念模型,并分析該過程固定和釋放砷的機理(圖6)。
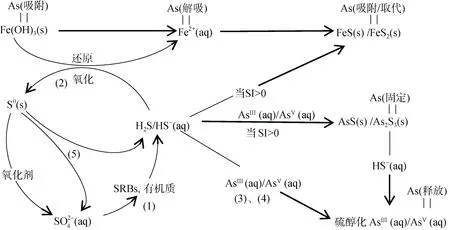
圖6 環境中鐵和硫氧化還原循環以及不同相之間的砷分配[65-69]Fig. 6 Fe and S redox cycle in the environment, and As partitioning among different phase
在存在乳酸鹽的硫酸鹽微環境中,初期存在硫酸鹽還原菌(SRBs)的微生物滯后期,然后硫酸鹽還原可以與Fe(III) 還原相當的速度進行,甚至由于動力學原因在某些階段取代Fe(III) 還原過程[65]。硫酸鹽被SRB 還原成硫化氫 (HS–和H2S 的混合物,pKa,H2S=7.02):SO+ 2C3H5O?HS-+ 2HCO+ 2C2H3O+H+(式1)。當鐵和砷硫化礦物達到飽和(即飽和指數> 0)時,游離硫化物(HS–)反應形成不溶性FeS 和砷硫化礦物(圖6),FeS 的形成可使土壤孔隙水中Fe(II) 濃度降低,而四方硫鐵礦的形成耦合As(V) 還原為As(III),As(III)進一步被As2S3類物質螯合[61]。如果存在活性Fe(III) 礦物質(如水鐵礦),則游離硫化物與其發生還原反應并產生Fe(II) 和零價硫(可能還有少量的硫代硫酸鹽):HS-+2Fe(OH)3(S)+ 5H+?2Fe2++S0(s)+ 6H2O(式2)。這個涉及電子轉移過程的反應由表面控制,是一個快速的化學過程,水鐵礦還原形成Fe(II),進一步轉化為針鐵礦和磁鐵礦[66],因表面積減少而導致砷從土壤沉積物中解吸。此外,砷的還原過程是砷與硫化物形成共沉淀的必要步驟[67],游離硫化物不僅可以還原活性Fe(III) 礦物質(式2),而且可以還原砷酸鹽[68]:H2AsO+ 3HS–+ 2H+?HAsS3O2–+3H2O(式3),HAsS3O2–+ HS–?AsS+ H2O(式4),通過形成溶解的硫醇化砷化合物如HAsS3O2–和AsSO3–4而增加砷的溶解度[69]。
零價硫的存在是硫酸鹽還原的診斷指標,因為它只能通過硫化物氧化形成(式1、式2)。然后零價硫可能會由微生物介導發生歧化反應,再次產生硫化物和硫酸鹽[67]:4S0(S)+4H2O?3HS–+ SO+ 5H+(式5)。
這一系列過程(式1、式2 和式5)形成了有效的生物地球化學硫循環,催化了Fe(III) 還原,并以相對低的水平緩沖溶解的硫化物濃度,直到活性Fe(III)被耗盡。該過程說明硫酸鹽還原產生的硫化物能夠還原Fe(III) 和As(V),促進砷釋放到水相[70],但又能通過促進砷和鐵硫化物礦物的沉淀而固定砷[71]。此外,不同介質產生的鐵水合物對砷的吸附能力顯著不同:由硫酸鹽培養基制備的鐵水合物具有高于硝酸鹽所制備的砷吸收容量[72],這說明了硫酸根離子能參與Fe(III) 水解,并且改變了所形成的水鐵礦的表面化學性質,使其更有利于砷酸鹽的吸附。另一方面,與吸附過程相比,As(V) 與Fe(III) 的共沉淀可產生更高的砷去除率,可能是因為在特異性吸附陰離子(即AsO)存在下吸附劑的活性位點和表面積最大化,并在共沉淀過程中形成了無定形的砷酸鐵使可溶性砷的量最小化,因此總體上有助于顯著降低砷溶解度。由此可見,微生物硫酸鹽的還原可以影響砷在土壤中的歸宿[61]。
如果硫循環過程中不存在活性Fe(III) 礦物質這種易氧化硫的氧化劑,則保持低活性的硫循環過程會被破壞,因此在相對還原的體系內(如填埋場),更有利于不溶性硫化物的形成和砷的固定。因此,關于密閉基巖含水層中硫化物氧化導致砷污染的報道并不多見,因為完全飽和的含水層中存在的硫化物由于還原環境和缺乏氧化劑而基本保持穩定。因此,厭氧條件下硫化物對砷的釋放遷移貢獻較小,但高砷含量的黃鐵礦和白鐵礦發生氧化則易導致砷的大量釋放。
2.2.2 磷酸鹽對砷的影響 盡管磷酸鹽(Pi)化合物經常被用于促進金屬如鉛和鋅的固化,降低其在土壤中的遷移率和生物利用度[73],然而在砷污染土壤中添加磷肥卻能增加砷的溶解度和流動性,促進植物對砷的吸收[74]。土壤中的砷主要以無機形式(砷酸鹽As(V)和亞砷酸鹽As(III))存在,有機形式的二甲基砷酸(DMAA)和一甲基砷酸(MMAA)的含量相對較小[75]。在有氧條件下,無機砷和Pi 由于相似的化學特性能夠形成穩定的+5 價氧化態四面體氧陰離子[76]。由于兩者物理和化學的相似性,As(V)和Pi 離子在相同土壤帶電表面存在強烈的相互競爭。在相同的Pi 和砷濃度下,由于Pi 陰離子具有更小的尺寸和更高的電荷,且不易與溶解的硫化物反應形成不溶的礦物質或溶液配合物,因此比As(V) 結合到土壤上更具優勢[77],從而更多的砷因吸附受到抑制而留存在溶液中被植物吸收。例如,Okkenhaug 等[78]發現,使用含磷的營養物質可以促進土壤溶液中砷的起始濃度由1 400 μg/L增大到1 700 μg/L;MorenoJiménez 等[79]也報道了,在Pi 存在的情況下增加了砷在土壤中的遷移。從另一角度,Tu 和Ma[80]發現添加磷可提高砷的生物利用度,施用磷對于使用蜈蚣草植物修復砷污染的土壤具有顯著的增強效果。此外,Pi 對土壤的吸附緩慢,隨著時間的推移將增加競爭As(V) 的吸附作用[76]。根據Steindorf-Rebhun-Sheintuch 方程、配體交換理論和共享電荷假設,Pi 有很大的可能取代土壤吸附的As(V)[81]。在使用飛灰改良的土壤中,Qafoku 等[82]報道了Pi 與砷酸鹽競爭全部可用的非特異性和特異性吸附位點,取代了As(III) 和As(V),增加了土壤中砷的流動性。
砷和Pi 的競爭吸附作用與土壤成分的性質有關,如砷酸鹽吸附量隨著土壤陰離子吸附量的增加而增加[83]。Violante 和Pigna[84]發現當As(V) 和Pi 以等摩爾比供應時,富鐵、錳礦物(如針鐵礦、綠脫石、含鐵綠土、水鈉錳礦和軟錳礦)吸收的As(V) 比Pi更多;然而無定形鋁礦物上(如三水鋁石、勃姆石、水鋁英石和黏土部分包括高嶺石、伊利石和蛭石等)吸附Pi 比As(V) 更多;對于任何pH 的石灰性紫色土壤,As(V) 和Pi 的吸附和解吸特性相似,遵循Langmuir 和Freundlich 方程[85]。在Bolan 等[83]的研究中,磷對砷的吸附和解吸影響取決于土壤陰離子的吸持能力,弱陰離子吸持能力的非水鋁英石土壤對砷的吸附量遠小于強陰離子吸持能力的水鋁英石土壤的砷吸附量,而磷的添加明顯促進了水鋁英石土壤中砷解吸,非水鋁英石土壤中添加磷對土壤砷的解吸和生物有效性的影響幾乎可以忽略。Peryea[86]也指出砷的解吸依賴于土壤類型,只有大量添加磷(P > 400 mg/kg)才會影響火山土壤中砷的溶解度。當土壤中Pi 含量非常高時,含陰離子土壤和堿性土壤中砷的遷移率和溶解度都會受到強烈的影響。
2.2.3 硅酸鹽和碳酸鹽對砷的影響 硅和碳是同族元素,硅酸根(SiO)和碳酸根(CO)對砷的競爭吸附機理相似。SiO具有與AsO3–3相似的空間結構,因此能夠同AsO競爭鐵礦物表面的吸附位點,促進SiO和HSiO從鐵礦物釋放。例如,Robins 等[87]指出,硅酸鹽能吸附在固體水鐵礦和鋁鐵礦的表面上,與Fe(III)發生絡合,從而導致As(V) 解吸;Swedlund和Webster[88]的研究也表明,硅酸鹽可抑制As(III) 和As(V) 在水鐵礦上的吸附。目前許多研究都忽略了碳酸鹽對吸附態砷酸鹽和亞砷酸鹽的取代作用,實際上地下水中碳酸鹽含量通常較高,且碳酸鹽吸附較強,可改變零電荷點(PZC)、Zeta 電位和氧化物的質子緩沖能力[89],能夠極大地影響砷的吸附。在自然環境下,吸附大量砷的沉積物存在于低濃度碳酸鹽的地表水中,當這些沉積物暴露于高含量的溶解碳酸鹽的地下水時,沉積物表面的砷就會因被CO取代而發生遷移。Appelo 等[90]使用表面絡合模型計算,證實了土壤和地下水中沉積物的吸附特別是碳酸鹽的吸附能夠顯著降低砷在水鐵礦上的吸附能力。這些研究說明硅酸鹽和碳酸鹽都是環境中砷濃度升高的促進因素。
2.3 土壤理化性質對砷遷移的影響
2.3.1 土壤氧化還原電位(Eh)對砷的影響 Eh 的周期性變化是水稻土獨特的特征,其變化范圍通常在+200 ~ +700 mV[91],這種廣泛的變化直接導致了水稻土中一些氧化還原敏感物質如氧、鐵、錳、氮、硫和碳的劇烈變化。淹水前,土壤孔隙結構可能充滿空氣,土壤環境的Eh 在300 mV 以上;淹水后,土壤孔隙網絡中的游離氧被微生物快速消耗,Eh 迅速降至200 mV 以下,此時微生物依次使用NO,Mn4+,Fe3+,SO作為電子受體進行還原,伴隨著微量氣體NH3,N2O,N2,H2S,CH4的排放以及pH 的上升[92],形成強烈還原的缺氧環境,土壤固相鐵礦物和As(V)均被還原溶解;大部分水稻收獲前一到兩周進行田間排水,Eh 又上升到400 mV 以上,還原態化合物如Fe(II) 和As(III) 被氧化Fe(III) 和As(V),As(V) 與鐵氧化物結合,含量及有效性均明顯降低[93]。
長期水淹條件會導致土壤Eh 降低,釋放到溶液中的 As(III) 和總砷的濃度均顯著提高。例如,Yamaguchi 等[94]指出當土壤Eh 從 +500 mV 降至+100 mV 時,土壤溶液中As(III) 濃度和Fe(II) 濃度顯著增高;Yamaguchi 等[95]發現,水淹條件下當水稻土的Eh 由+100 mV 下降至 –68 mV 與 –75 mV 時,溶液中砷的釋放量每千克土分別提高了6.9 μmol 和19 μmol;Syu 等[96]報道了砷污染的水稻土經歷淹水后土壤溶液砷含量大幅度上升至3 000 μg/L 以上,伴隨著Eh 的急劇下降與Fe(II) 含量的快速上升;進一步研究指出砷濃度與Eh 存在關系可表示為[As]=5.84exp(–0.0145 Eh)[16]。這些研究說明土壤砷的釋放量和土壤Eh 值呈顯著負相關關系。然而,當淹水70 d 后,水稻土壤溶液中的砷濃度趨于保持穩定[97],可能是由于長期淹水使水稻土的pH 保持相對穩定的動態平衡和氧化還原態平衡,從而砷也保持在土壤和水之間的分配平衡。此外,反復氧化還原循環可以降低還原過程中高達45% 的砷遷移率[98],其原因可能是Eh升高時形成的次生鐵礦物α-FeOOH/Fe(OH)3與砷共沉淀。由此可見,土壤經歷淹水缺氧過程導致Eh 下降,砷的遷移率增加,但長期淹水或反復淹水可能保持一定的As(III) 含量不變甚至降低其遷移率。
2.3.2 土壤pH 對砷的影響 土壤pH 是影響砷形態和有效性的關鍵理化性質之一。厭氧條件下由于微生物對有機質的降解可導致pH 降低,這種酸性條件有利于礦物表面的質子化、砷酸鹽陰離子(H2AsO–4或HAsO2–4)的吸附以及降低As(V) 的溶解度,提高亞砷酸(H3AsO3)的溶解度。在堿性條件下,As(III) 優先于As(V) 吸附在氧化鐵表面上;在近中性的pH,即接近H3AsO3的pKa(9.2),砷酸鹽和亞砷酸鹽都通過形成強內層絡合物在水合氧化鐵(HFO)和結晶氧化鐵的表面處以相同的效率吸附[99]。Honma 等[16]研究表明,溶解性砷濃度與pH 存在關系可表示為[As]=3.56×10–12exp(4.72 pH);相似的研究也指出總砷和pH的關系式為[As]= –0.557+3.61×10–11exp(3.50 pH)[15],表明土壤砷的釋放量和土壤pH 呈正相關。這些研究說明了水稻土微環境中Eh 降低和pH 升高是驅動砷釋放的關鍵因素。
2.3.3 有機質對砷的影響 土壤有機質是鐵代謝微生物和礦物質相互作用過程的重要參與者,它的氧化能夠直接耦合土壤沉積物和溶液中砷的微生物還原[100]。有機化合物尤其是可提供離子參與形成Fe(III)絡合物的有機物,會嚴重干擾砷酸鹽在水鐵礦和相關吸附劑上的吸附,如黃腐酸(FA)或腐殖酸(HA)吸附在針鐵礦表面可導致砷酸鹽的解吸,增加溶液中砷濃度[101]。一方面,有機質中的羧基(–COOH)和羥基(–OH)官能團能夠與金屬氧化物發生配位體交換[78],提高土壤溶液中砷的有效性[21];另一方面,有機質的微生物降解通常耦合Fe(III) 礦物還原過程,進而改變土壤固相或土壤溶液的平衡狀況,促進土壤固相中砷的解吸[21]。Fe(III)和As(V) 同為電子受體,二者之間可能存在競爭還原與氧化關系,在足夠電子供體情況下鐵砷先后被還原,但在電子供體不足的情況下,氧化還原過程中鐵和砷之間可能存在競爭,如圖7 所示。
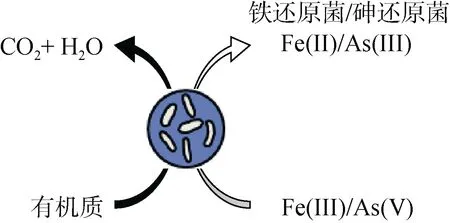
圖7 有機質參與砷還原過程Fig. 7 Organic matter involved in As reduction process
有機陰離子促進As(V) 釋放能力大小為檸檬酸鹽>草酸鹽>>乙酸鹽[102],環境中競爭性物質對砷吸附的影響順序為磷酸鹽>腐殖酸>硅酸鹽>硫酸鹽[103],其干擾作用(尤其是腐殖酸)在較堿性pH 時增加。腐殖酸和磷酸鹽、硅酸鹽不僅通過競爭性吸附降低砷的吸附,還能與Fe(III) 氫氧化物形成復合物,而硫酸鹽主要通過競爭性吸附抑制砷的吸附,因此前者抑制砷的吸附作用比硫酸鹽更強。此外,通過提供更高的電子供體(如乳酸鹽)濃度可提高Fe(III) (氫)氧化物還原速率,從而加速As(III) 的解吸[12]。由此可見,有機質也是影響砷還原釋放的關鍵因素之一。
值得注意的是,砷的釋放程度并不總是與固體表面吸附其他物質的量相匹配,因而砷的釋放不能僅僅用競爭吸附/解吸來解釋。砷的解吸應通過砷的競爭吸附/解吸、鐵和鋁的氧化/氫氧化物的溶解以及Fe(III)和Al(III) 與As(V)、OH–、檸檬酸鹽、草酸鹽、硅酸鹽、磷酸鹽、以及pH 和Eh 等多種因素的復合來解釋砷解吸的復雜機制。
3 結論與展望
砷具有獨特的物理和化學性質,在環境中的遷移過程是通過生物地球化學反應與人類相互作用的復雜結合而發生的,受微生物、鐵、鋁、錳、硫、磷、硅酸鹽、碳酸鹽、pH、Eh 和有機質等諸多因素的影響。對文獻的分析表明,造成砷向環境遷移的主要原因是鐵錳氧化物礦物的還原溶解、黃鐵礦等硫化物礦物的氧化溶解、鋁氫氧化物表面的砷還原、有機質抑制砷吸附以及Eh 降低和pH 升高。未來的對砷深入研究應集中在下列3 個方面:
1)探討上述因素在不同性質土壤中的相互作用以及促進砷釋放的相對貢獻值。砷的釋放可能通過上述多種因素的復合,作為pH 和Eh 的函數來表達砷解吸的復雜機制;應重點考慮鐵錳氧化物以及碳酸鹽,目前有關錳氧化物對污染土壤中As(V) 還原和釋放影響的研究較少,碳酸鹽是研究砷時最易忽略的一個因素,而地下水中碳酸鹽含量通常較高且對砷的吸附影響作用較大。此外,砷在全球土壤時空分布規律以及導致當地高砷含量的因素、降低當地砷污染的對策有待進一步研究;目前已有許多研究報道了當地地下水砷的分布規律及遷移釋放的影響因素,然而對于這方面內容在全球地下水中缺乏綜合性分析。
2)進一步研究厭氧條件下鐵砷還原與氧化過程的動力學機制以及鐵砷耦合的主要影響因素。目前有關鐵參與砷的形態變化和遷移轉化研究較多,然而控制鐵還原過程的關鍵因素仍不清楚。Fe(III) 與 As(V)同為電子受體,兩者間的競爭性也缺乏研究。研究證實硝酸鹽依賴性細菌 FeOBAcidovoraxsp. BoFeN1在添加 As(III) 的時候,系統中的鐵礦物被還原,說明在微生物體內確實存在鐵砷耦合的反應機制,然而微生物在鐵砷耦合作用過程中的生物化學機理有待進一步研究。
3)土壤中砷的氧化還原轉化與微生物息息相關,砷氧化微生物可分為化能自養型砷氧化微生物(CAOs)、異養型砷氧化微生物(HAOs)兩類,CAOs 在厭氧環境中由硝酸根作為電子受體。HAOs 同樣也能夠氧化As(III),但是需要有機物質作為能量與細胞物質的來源。目前,砷代謝微生物研究主要集中于可培養菌群對砷的遷移轉化的影響及其機理的研究,環境中不可培養的砷代謝微生物的生物多樣性和群落結構有待進一步的研究。
猜你喜歡
體育科技文獻通報(2022年3期)2022-05-23 13:46:54
天津外國語大學學報(2021年3期)2021-08-13 08:32:18
遼金歷史與考古(2021年0期)2021-07-29 01:06:54
科技傳播(2019年22期)2020-01-14 03:06:54
遼金歷史與考古(2019年0期)2020-01-06 07:45:20
民用飛機設計與研究(2019年4期)2019-05-21 07:21:24
電子制作(2018年11期)2018-08-04 03:26:04
汽車工程學報(2017年2期)2017-07-05 08:13:02
國際商務財會(2017年8期)2017-06-21 06:14:14
電子制作(2017年23期)2017-02-02 07:17:19
