原料藥企業藥品GMP認證檢查缺陷分析
2020-05-25 02:29:16李想高永寶王璐王曉魏晶
品牌與標準化 2020年3期
李想 高永寶 王璐 王曉 魏晶
【摘要】 研究目的:通過對遼寧省原料藥生產企業GMP認證檢查中發現缺陷的分析,推進原料藥企業對GMP的理解,提高企業的管理水平。主要方法:對2018年至2019年遼寧省原料藥企業藥品GMP認證檢查中發現的缺陷進行統計分析,對主要問題進行分類總結。結果與結論:通過對缺陷進行分析發現,原料藥企業在質量控制與質量保證、文件管理、附錄等方面的管理較為薄弱,建議原料藥生產企業務實人員培訓,做好物料管控、確認與驗證及文件管理工作,重視變更和偏差管理,提高質量意識。
【關鍵詞】 原料藥;GMP認證;高頻缺陷
Abstract: Objective: Through the analysis of the defects found in the Good Manufacture Practice(GMP) certification inspection of active pharmaceutical ingredients(API) manufacturers in Liaoning Province , promote the understanding of GMP by manufacturers.. Methods:Statistical analysis was performed on the defects found in the GMP certification inspection of Liaoning Province from 2018 to 2019, and the defect items in the GMP certification inspection were classified and summarized.Results and Conclusion: API manufacturers are weak in quality control and quality assurance, document management, appendices, etc., ., it is recommended to implement the training of operators in every personnel; strengthen the materials supervision; improve the confirmation and verification; pay attention to details and do a good job of confirmation and verification; systemize document management and standardize record filling; attach importance to change and deviation management, and improve quality awareness.
Key words: active pharmaceutical ingredients;GMP certification;main defects
本文對2018年至2019年遼寧省原料藥企業藥品GMP認證檢查工作中發現的高頻缺陷進行針對性的分析,并提出相應的改進措施和建議,為原料藥企業GMP實施的規范性提供參考。
1 認證檢查情況概述
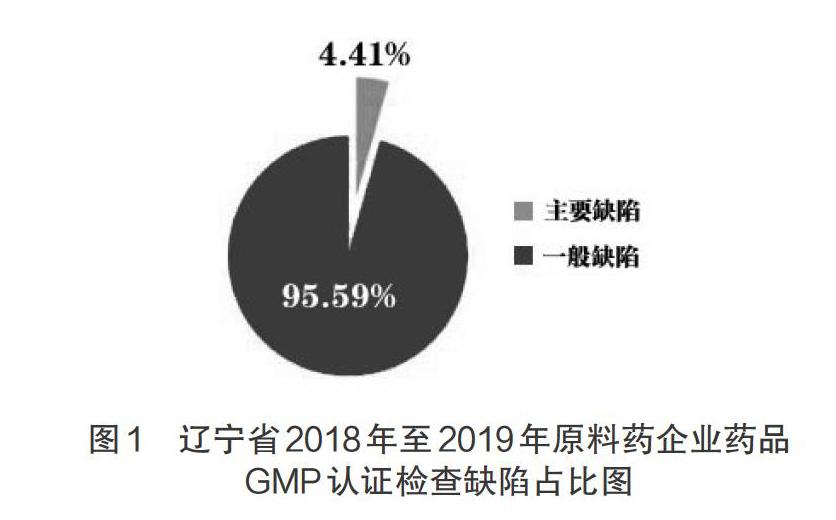
2018年至2019年,遼寧省認證審評院共接收原料藥企業新修訂藥品GMP認證申請16家次。發現缺陷項目總計136項,其中嚴重缺陷0項、主要缺陷6項,一般缺陷130項,主要缺陷及一般缺陷分別占總缺陷的4.4%、95.6%;具體見圖1。
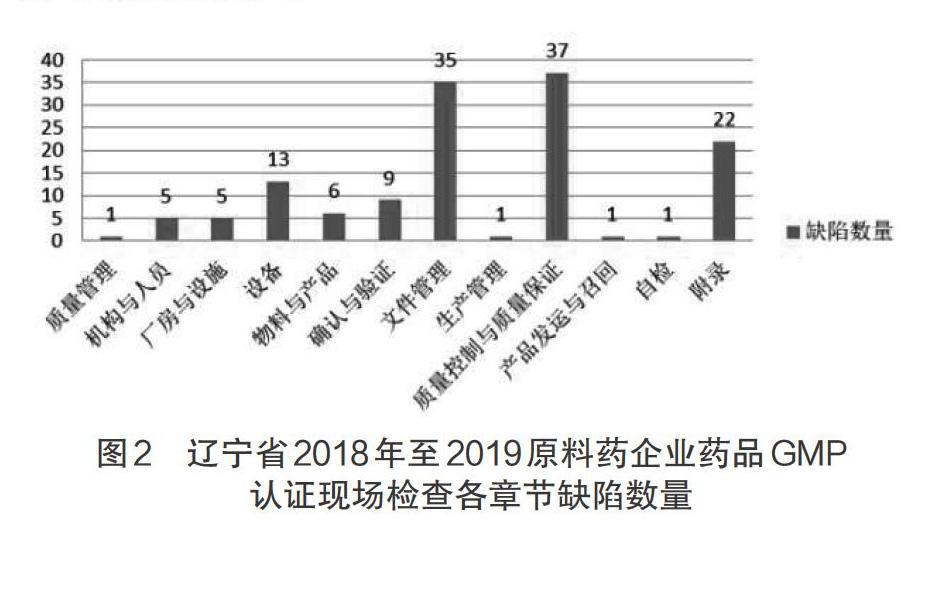
缺陷數量居前三位的章節分別為質量控制與質量保證、文件管理、其他(含原料藥、計算機化系統、確認與驗證附錄),數量分別為37項、35項以及22項,分布情況見圖2。
認證現場檢查各章節缺陷數量
2 缺陷項目描述與分析
2.1 質量控制與質量保證部分
2.1.1 質量控制方面
質量控制實驗室的相關記錄不規范或記錄不全面,典型缺陷舉例如下:
1)某品種成品批檢驗記錄中【鑒別】項紅外圖譜中未體現采集時間信息。
2)0.1 mol/L硫代硫酸鈉滴定液配制記錄未按《滴定液管理規定》進行F值計算。
3)某原料成品批檢驗記錄中,含量測定項下未設置樣品和對照品的具體稀釋步驟及對照品稱量項目。
4)部分批檢驗記錄記載的信息不全,如:熔點測定項未記錄樣品的制備過程;含量測定項未記錄實驗室溫度。
5)對氮氣貯罐的氮氣定期監測含氧量,并通過氧含量計算氮氣純度,此計算方法不準確。
2.1.2 穩定性考察存在不足
1)長期實驗留樣某品種臺賬顯示剩余12瓶,實際檢查中發現剩余6瓶,與臺賬不符;
2)《某原料中間產品穩定性考察方案》中,中間體1及中間體2的穩定性考察周期均為18天,但工藝規程規定中間體1暫存時限為1個月,中間體2暫存時限為12個月,缺乏數據支持。
2.1.3 質量保證方面
2.1.3.1 規程不能指導實際操作
1)輔料蔗糖比旋度測定未注明取樣量和操作過程;
2)紅外鑒別項未附紅外標準圖譜;
3)企業未建立關于修改積分參數和進行手動積分的色譜系統積分操作規程;
4)企業《化工原料取樣操作程序》未規定在風險評估的基礎上選取有代表性的樣品進行取樣(例如,未考慮運輸途中的最差條件)。
2.1.3.2 變更、偏差管理不到位
1)物料供應商發生變化時,企業進行了供應商審計和評估批準,但質量標準中檢驗指標發生變化時,企業內控標準未調整。如:甲胺鹽酸鹽,供應商為常州某所,內控標準:未設干燥失重檢測項,生產廠家檢驗報告單中有該檢測項目;
2)精餾塔加熱方式由導熱油加熱變更電加熱,個別文件未及時變更,如:某車間精餾系統確認文件中未包含精餾塔電加熱確認內容;
3)《偏差和糾正預防措施管理規程》未對重復出現的偏差如何處理進行規定。
2.1.3.3 供應商管理方面存在不足
1)參與反應的關鍵物料未作為主要物料納入現場審計范圍;
2)企業確定的合格供應商中,部分供應商長期未發生購進業務,但企業管理文件中未明確此類供應商的審計、評估方式;
3)企業供應商審計人員缺少化工專業知識的培訓。
2.1.3.4 缺陷分析
企業首先要加強質量控制與質量保證意識,不論質量控制實驗室還是車間生產,文件的起草、審核、批準等管理過程及實施過程中詳盡的記錄,目的是為了保證原料藥生產過程的質量可控性和可追溯性。企業應確保所有人員正確理解并執行生產工藝、質量標準、檢驗方法,避免偏差的產生,對已經發生的偏差按程序報告、記錄、調查、處理并采取糾正措施和預防措施。
2.2 附錄部分
在新修訂藥品GMP正文的基礎上,國家食品藥品監督管理總局陸續發布了《確認與驗證》、《計算機化系統》等附錄,完善了GMP的架構,細化了質量管理的要求。原料藥生產企業由于起始物料多、生產工藝比較復雜,附錄有針對性的提出了更詳細、更深層的執行標準,是對原料藥生產企業的質量管理的挑戰,也是原料藥企業生產管理和質量管理的薄弱環節。在近兩年對原料藥企業的檢查中,原料藥附錄、計算機化系統附錄、確認與驗證附錄中發現的缺陷項目共計22項,占一般缺陷項目總數的25.7%。
2.2.1 原料藥附錄方面
2.2.1.1 雜質檔案、溶媒回收方面
1)企業建立了回收溶劑的內控質量標準并定期檢驗,但未建立原料藥的雜質檔案;
2)生產過程中對回收丙酮和回收正己烷的密度進行控制,但無使用周期或最多使用批次的相關規定。
2.2.1.2 清潔驗證、防止交叉污染方面
1)《原料車間多品種共線生產質量風險評估》中描述“共線生產的品種彼此間不存在反應或毒性”的結論缺乏科學依據,另報告中缺少共線品種防止交叉污染的措施評估;
2)二維混合機清潔驗證方案中未指明清潔參照物;
3)從正沸接收罐放料口用敞口的鋁桶接收成品,無避免污染的措施。
2.2.1.3 文件規定與實際不一致
1)《某原料藥工藝規程》未規定0.45 μm、0.22 μm過濾器的使用次數、壓力等相關要求;現場檢查時某原料藥加甲醇時即開啟氮氣保護,與工藝規程規定的加入精左鹽完全溶解后開啟氮氣保護不一致;
2)某原料藥工藝規程中描述的部分工序的工藝參數描述不具體,例如:“粗品精制工序規定將精制罐內物料冷卻至5 ℃以下結晶至少0.5小時”,未規定溫度和時間范圍。
2.2.1.4 缺陷分析
遼寧省原料藥生產企業在原料藥附錄執行過程中存在的缺陷主要表現在工藝規程描述不具體、清潔驗證不規范、雜質檔案未建立、溶媒回收使用次數未規定等方面。工藝規程描述的不具體會造成操作規程及相關批記錄上的內容存在相應問題,對產品的質量控制和質量保證存在風險,應加強管理;原料藥生產企業使用的溶媒可以回收套用,但是需要針對其回收使用的次數進行驗證,方可保證產品的質量;原料藥生產企業設備的清潔驗證方面的概念還不夠清晰。清潔驗證方案應該明確清潔目標,即:在哪些生產設備中,殘留物是什么,殘留物的水平、清潔方法、清潔標準、取樣方法、分析方法都需要描述清楚;驗證報告中要有清洗樣品測試結果及其結果分析和匯集,清洗執行和分析檢測的偏差都需要解釋并說明原因,檢測清潔后殘留物量的分析方法應經過驗證。
2.2.2 計算機化系統附錄
2.2.2.1 計算機化系統文件制定不全面
1)《計算機化系統管理規程》未對刪除或棄用數據進行相關規定;
2)《成品運輸確認報告》中未對冷鏈物流公司的溫控數據進行收集匯總。
2.2.2.2 權限及相關設置不清晰
1)企業質量負責人沒有使用和管理計算機化系統(審計追蹤功能)的職責權限;
2)《化驗室計算機化系統權限賬戶密碼管理及電子數據備份管理》未規定紙質版和電子版哪個是主數據。
2.2.2.3 缺陷分析
計算機化系統作為藥品質量風險管理的一部分,風險管理應該貫穿計算機化系統的生命周期全過程;計算機化系統的驗證是計算機化系統管理的基礎,其驗證包括應用程序的驗證和基礎構架的確認,其科學的風險評估應當充分考慮計算機化系統的適用范圍和用途,且在計算機化系統的生命周期內保持其驗證狀態;通過驗證制定相應的操作規程并對相關的人員進行培訓;計算機化系統的文件管理應該遵從GMP文件管理程序,文件的變更應該得到及時的審核、批準;記錄應該具有可追溯性,且真實可靠。對于原料藥企業來說,計算機化系統方面的缺陷多數體現在計算機化系統的驗證、數據的審計追蹤,數據完整性及文件管理等方面的問題。企業應當根據實際生產和檢驗過程中使用到的計算機化系統進行分類和評估,確保所采取的措施復核GMP的規定。
2.2.3 確認與驗證附錄
2.2.3.1 驗證數據不充分
確認與驗證部分缺陷主要表現在驗證數據不充分,典型缺陷如下:
1)《離心機性能確認》中未明確干燥失重限度標準;
2)企業未按照《驗證管理SOP》規定,按期開展品種工藝再驗證;
3)工藝驗證方案部分參數未設置,如:未設置酰化滴加溫度。
2.2.3.2 缺陷分析
確認與驗證是GMP的重要組成部分,部分企業驗證管理不規范,不能正確理解確認與驗證的意義。原料藥生產設備驗證與工藝驗證是原料藥生產企業執行GMP的核心部分,工藝參數、工藝規程、生產操作規程及批生產記錄等的制定均依據驗證的結論結果,充分的驗證可以減少或避免原料藥生產過程中產生的偏差,保證藥品質量持續穩定。應采取風險評估的方式確定工藝驗證方案,工藝驗證執行中做好記錄,如驗證過程中出現偏差要依據相應偏差管理規程進行偏差處理,最終確定準確的工藝參數范圍。
2.3 文件管理部分
從近兩年GMP實施情況看,原料藥生產企業“重生產、輕質量”的情況得到一定改觀。企業基本已經建立質量管理體系并初步運行,但是仍有一部分企業的文件管理水平存在缺陷,主要體現在文件制定脫離生產實際、不按照文件執行等問題。在文件管理環節共計發現35項一般缺陷,占一般缺陷總數16.2%。
2.3.1 規程類文件不能指導實際生產活動
1)生產操作規程中規定:反應10-11小時,補加新鮮的A物料10-20公斤,繼續反應,反應溫度又逐漸降低,此操作約2小時一次,共補加新鮮的A物料120公斤,是否需要移出生成的低沸物,描述不清晰;補加的120公斤的A物料是否包括第一次補加A物料,描述的不清晰;
2)生產工藝中部分需要調控溫度的操作,在工藝規程表述中方式不清晰、確切,如降溫到某某度表述為降溫某某度;
3)工藝規程表述不到位,指導不明確,如成品批(商品批)為多亞批總混而成,但工藝規程中顯示的處方量為每亞批原料用量,亞批的重復操作未在規程中表述(未發現實際生產不符合)。
2.3.2 記錄內容不全面,缺少主要關鍵項目
2.3.2.1 未及時填寫記錄或記錄填寫錯誤
1)原料批檢驗記錄中含量測定項公式錯誤,未按無水物計算含量(含量測定結果符合規定);
2)《成鹽崗位操作記錄》缺少加入活性炭后壓濾器檢漏過程的記錄;冷卻析出和開啟攪拌的順序描述有誤;
3)《***成鹽崗位操作記錄》缺少對溶解結果的判斷記錄。
2.3.2.2 批生產記錄不規范
1)批記錄未記錄所用原料的化驗單,無設備清潔合格證;
2)精制乙酸乙酯只記錄了蒸餾時間、溫度,未記錄真空度;蒸餾終點判斷不明確,只描述在-0.06~0.09 mpa真空度下減壓蒸餾。
2.3.2.3 缺陷分析
文件管理方面的缺陷主要表現在工藝規程、操作規程、年度質量回顧分析報告等管理規程以及相關記錄上,文件規定或描述的內容不完整或不符合規范要求,不能確保質量管理系統的有效性,新修訂藥品GMP強化了文件的管理,目的在于保證文件的合法性、權威性、系統性、有效性。應加強文件規范管理以確保質量控制和質量保證方面的管理更加完善。
3 提升原料藥生產企業GMP管理水平的建議
做好原料藥生產企業的GMP實施工作,是一項長期而具有挑戰性的工作。通過對上文缺陷項目的總結分析,可以發現原料藥生產企業在GMP實施中存在的某些共性問題,針對這些共性問題,結合原料藥的自身特性,本文提出了以下建議,以提升企業的管理水平。
3.1 加強人員培訓,提升人員素質
實施藥品GMP,人員是關鍵因素。GMP的各個環節均需要“人員”的參與與執行。原料藥的生產與其他的藥物制劑生產有本質上的區別,原料藥生產其實是一個化學反應過程,化學反應中起始物料、環境以及設備的差異都會直接對產品質量和收率產生影響。如滴加、萃取、濃縮等操作更多依靠的是人員對反應溫度、速率、現象、終點的判斷[1]。而這種判斷是建立在長期有針對性的培訓而形成的一種經驗。因此,應加強對操作人員培訓,并注重培訓效果。
針對原料藥生產企業培訓工作應著重于以下幾點:1)加強人員管理,專人進行培訓。,培訓人員需熟悉每種原料藥產品特性,制定不同崗位、不同層次有針對性的培訓計劃,嚴格落實培訓制度與培訓內容;2)培訓內容應全面且有針對性。內容應包括合成工藝、化工物料等方面的專業知識;3)落實培訓效果,量化考核指標。應將培訓作為員工主要工作內容之一,并體現在績效考核中,并將培訓資料以月度或季度為周期進行歸檔留存[2]。
3.2 重視變更和偏差管理,提高質量意識
原料藥生產工藝十分復雜,少則幾步,多則幾十步,關鍵工序的質量控制與雜質的種類與數量密切相關,另外生產設備及關鍵參數也對原料藥質量產生至關重要的影響。隨著制藥行業的不斷發展,制藥設備的不斷更新,企業需要結合品種的不同特點,實時關注生產過程中的各種異常情況,積極調查與應對,不斷累積數據,用以提升過程控制能力與產品質量。應加強變更控制、偏差管理、超標調查等方面的培訓。質量管理部門應加強對檢驗數據和檢驗記錄的復核,出現偏差迅速進行偏差處理,并且對檢驗記錄進行全過程審核,包括對照品的批號、來源、純度及計算方法審核。
3.3 文件管理系統化,記錄填寫規范化
文件是質量保證系統的基礎要素,是藥品生產企業實施GMP的依據與行為準則。一個有效的文件管理體系可以分為四級文件:一級文件為質量方針與質量目標,二級文件為質量管理文件,三級文件為操作規程,四級文件為質量記錄。要形成規范的文件管理體系,必須明確各個部門職責和崗位職責,配備有經驗的生產和質量管理人員,統一協調文件的編制、修訂、批準,形成長效機制,才能逐步建立有效的文件管理體系[3]。企業應當按照生產品種的特點,形成有實際指導意義的文件體系,做到“記你所做的,做你所寫的”,真正的將文件體系與實際生產相結合,不斷在實踐中提升和優化文件體系[4]。
3.4 注重細節,有針對性做好確認與驗證工作
驗證一直被認為是藥品生產企業實施GMP認證過程之中的薄弱環節[5]。近年來由于藥品GMP的實施。各類指南的發布,極大地提高了原料藥驗證管理的水平。但是結合目前的檢查情況來看,原料藥生產企業還應從以下幾個方面加強驗證管理:1)充分認識到驗證工作的重要性,根據生產和質量管理的實際情況科學制定驗證計劃、編制驗證方案,同時重視原始數據的搜集、分析和評價以及驗證工藝的偏差處理,做好驗證文件的歸檔保存[6]。2)除了廠房設施、生產工藝、分析方法等日常驗證工作之外,還應結合原料藥生產實際,有針對性的開展驗證工作。如溶劑回收使用,應充分評估溶劑回收的用途,制定合適的質量標準、監控回收溶劑中雜質的含量,通過驗證確認回收方法與回收溶劑用于原料藥生產的可能性[7]。
3.5 提升安全管理意識,貫徹執行生產全過程管理
原料藥生產使用的起始物料多數為化工原料,大部分具有一定的毒性;部分普通原料藥的生產批量較大,使用的起始物料種類多,數量大;部分工序涉及的反應為放熱反應,風險系數較高,建議加強安全管理,提升風險意識,定期開展安全檢查工作,對風險系數高的危險操作實行全面監控,不斷強化操作人員防范意識,嚴格按照工藝規程進行原料藥生產[8,9]。不僅保證所生產產品的安全性、有效性,更要保證生產環節的安全性。
4 結語
根據新版的藥品管理法,藥品GMP認證這種許可形式已經退出歷史舞臺,但是藥品認證檢查從來不是目的,而是有效保證藥品質量的一種手段。伴隨GMP的不斷發展,原料藥生產企業應持續結合自身的實際情況,規范各項藥品生產管理活動,確保企業能夠始終如一的生產出安全有效的產品。
【參考文獻】
[1] 胡剛.從事原料藥生產管理需要重視的五個因素[J].中國藥物經濟學,2012(5):227-229.
[2] 王小衛.淺談新版GMP認證藥品生產企業的培訓管理[J].廣州化工,2013(17):263-264.
[3] 茌艷艷.如何提升產品質量檢測機構質量管理體系有效性[J].商品與質量,2017(3):143.
[4] 孫煜,呂思伊,魏曼.湖北省原料藥GMP認證中發現的主要缺陷及建議[J].中國藥事,2018(9):1264-1270.
[5] 高文昊.原料藥實施新版藥品GMP缺陷情況分析[J].民營高文昊.原料藥實施新版藥品GMP缺陷情況分析[J].民營科技,2018(8):11.
[6] 王聞珠.執行新修訂的《藥品GMP認證檢查評定標準》應重視的幾個問題[J].中國藥事,2008(8):649-651+674.
[7] 王璐,趙紅菊,馬輝,等.遼寧省藥品GMP認證檢查缺陷情況分析[J].中國藥事,2017(5):520-523.
[8] 徐占武.原料藥生產過程風險管理研究[J].鹽科學與化工,2018(3):21-23.
[9] 陳慧萍.質量風險管理在藥品GMP認證檢查中的應用[J].中國藥品標準,2010(6):411-413.
【作者簡介】
李想(1994-),女,碩士研究生,研究方向為藥事管理學;高永寶(1966-),男,高級工程師,學士,研究方向為藥物合成;王璐(1985-),女,副主任藥師,碩士,研究方向為藥物制劑;王曉(1986-),男,主管藥師,碩士,研究方向為藥事管理與法規。
通訊作者:魏晶(1963-),女,教授,碩士研究生導師,博士,研究方向為藥品、醫療器技術監管研究與安全性評價。
