二茂鐵類衍生物對HAN/PVA熱分解影響研究
2020-05-13 12:15:52胡松啟劉林林車琪銘劉元敏
火炸藥學報 2020年2期
胡松啟,康 博,張 研,劉林林,車琪銘,劉元敏
(1.西北工業大學燃燒、熱結構與內流場重點實驗室,陜西 西安 710072;2.西安航天動力技術研究所,陜西 西安 710025)
引 言
微推進系統能夠實現微小航天器的姿態調整、軌道控制和重力補償等,固體化學微推進系統以固體推進劑為能源,具有結構簡單、體積小和質量輕等優點,應用前景廣闊[1-2]。但常規固體推進劑用于制備微推進劑藥柱存在燃燒效率較低和生產安全性差等方面的問題,難以滿足微推進系統的裝藥要求。硝酸羥銨(HAN)極易溶于水,采用HAN水溶液作氧化劑更易于實現微藥柱的制備,且微藥柱的燃燒受原材料的尺度效應影響小,有望實現高效燃燒,是一種較為理想的微固體推進劑用氧化劑。考慮到水溶性聚乙烯醇(PVA)常與HAN溶液配合作為凝膠推進劑和電控推進劑的黏合劑,故可采用PVA作為HAN基微固體推進劑的黏合劑[3-5]。
然而,硝酸羥銨/聚乙烯醇(HAN/PVA)推進劑在制備過程中PVA與水發生溶脹,從而水將氧化劑與燃料隔絕開來,且黏合劑的反應活性低,皆可能導致推進劑點火和燃燒困難。在推進劑點火過程中,點火藥產生的燃氣使推進劑燃面升溫而發生熱分解,分解產物進行強烈的氧化還原放熱反應,產生火焰并最終實現推進劑的點燃。火焰向推進劑內部傳播的過程即燃燒過程[6]。因此推進劑的點火燃燒起始于熱分解,若對推進劑的熱分解過程進行有效催化將有助于其點火燃燒性能的提升。
深入認識HAN的熱分解機理是開展HAN/PVA推進劑熱分解催化研究的基礎。目前一般認為固體HAN的分解為質子傳遞反應,起始分解溫度約為453K;HAN溶液的熱分解為自催化過程,其分解會受到水的初始濃度影響;HAN的分解產物包括N2O、NO等氧化性氣體,NO2是一種主要的中間產物[7-10]。在HAN基推進劑催化方面,通常采用銥和鉑等貴金屬來催化點火,但需要將催化床預熱至473K,低溫下點火延遲期長甚至無法點火,且推進劑較多積存在催化床上,導致發動機的啟動性能受到限制。Popa、Kappenstein和Courthéoux等[11-14]在HAN催化分解的基礎研究中取得了重要進展,采用溶膠-凝膠法和二氧化碳超臨界干燥法制備了基于Al2O3的載體和催化劑,并引入質量分數5%的Pt金屬,能夠實現HAN水溶液在313K以下催化分解。此外,HAN基推進劑和催化劑匹配性問題也十分重要,匹配性不佳的配方會產生催化床空腔、催化劑壽命下降和試車穩定性差等問題,影響發動機的使用。
胡建新等[15]在HAN/PVA電控推進劑的研制過程中,加入二茂鐵及其衍生物來提高推進劑的燃速,但這類催化劑對推進劑熱分解特性的影響規律卻未見報道,考慮到推進劑的點火燃燒起始于熱分解,因此本研究選擇二茂鐵類衍生物作催化劑,采用熱分析儀對HAN/PVA的熱分解機理和動力學參數進行分析討論,研究結果可為解決HAN/PVA的點火和燃燒困難問題提供幫助。
1 實 驗
1.1 試劑與儀器
HAN水溶液,質量分數80%,通過復分解法自制。PVA,化學純,醇解度99.8%~100%,分子質量89000~98000,天津市科密歐化學試劑有限公司;卡托辛(Catocene)、乙基二茂鐵(Ethylferrocene)、辛基二茂鐵(Octylferrocene)、叔丁基二茂鐵(t-Butylferrocene),營口天元化工研究所股份有限公司。實驗樣品組成及二茂鐵類衍生物主要理化性能見表1。
TGA/DSC1同步熱分析儀,瑞士梅特勒-托利多公司;Tensor27紅外光譜儀,德國布魯克光譜儀器公司;OMNI star質譜儀,德國皮埃爾真空儀器公司。
1.2 HAN/PVA樣品制備
通過熱力學計算可知,在燃燒室壓強為5MPa、壓強比pc/pe為50的條件下,當HAN水溶液和PVA質量比為87∶13時,推進劑的理論比沖最高,選用該配比本實驗制備了5個樣品,制備過程為:首先將HAN水溶液和PVA混合,在338.2K下攪拌1~2h,直至藥漿中無PVA微小顆粒,在室溫條件下將質量分數為1%的二茂鐵類衍生物加入樣品1~樣品4中并充分攪拌;然后將藥漿在338.2K下真空熟化1~2d后將溫度降至308.2K成型,制得HAN/PVA樣品。樣品1~樣品5的組成見表1。
注:w為質量分數;P為純度。
1.3 熱分析實驗
本研究首先采用TGA-DSC-FIRT-MS聯用儀對22.8mg的HAN/PVA樣品進行測試,測試氛圍為氬氣,溫度范圍為313.2~973.2K,升溫速率為10K/min。然后利用TGA/DSC同步熱分析儀,對樣品1~樣品5分別在升溫速率7、10、15和25K/min條件下進行熱分析測試,試樣質量為(3 ± 0.1)mg,溫度范圍為308.2~623.2K。
2 結果與討論
2.1 HAN/PVA熱分解過程
升溫速率為10K/min時,HAN/PVA樣品的TGA-DTG測試結果如圖1所示。HAN/PVA樣品在升溫速率7、10、15和25K/min條件下測得的DSC曲線如圖2所示。
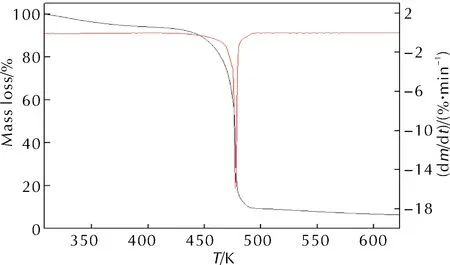
圖1 HAN/PVA樣品的TGA-DTG曲線
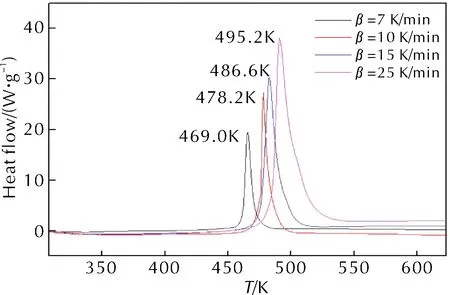
圖2 不同升溫速率下HAN/PVA樣品的DSC曲線
由圖1和圖2中10K/min的DTG和DSC結果可知,HAN/PVA樣品熱分解僅有一個明顯的放熱分解峰,起始分解溫度為457.0K,峰值溫度為478.2K,終止分解溫度為485.2K,由此推測HAN/PVA的熱分解過程是一個固態分解過程,在此過程中樣品的質量迅速降低,失重率約為73.7%,放熱量約為1312.1J/g。由于PVA高分子的熱分解通常發生在533~553K左右,因此該階段的失重由HAN的熱分解速度控制,快速失重和尖銳放熱峰表明HAN在短時間內放出了大量的熱。由圖2可知,隨著升溫速率加快,HAN/PVA樣品的起始分解溫度和終止分解溫度均升高,DSC曲線上的分解峰值溫度升高,峰面積也變大,即放熱量增大。
結合TGA-DSC-FIRT-MS聯用儀測得的紅外光譜和質譜結果,進一步研究HAN/PVA的熱分解過程。不同溫度下HAN/PVA熱分解逸出氣體產物的紅外光譜圖和質譜圖如圖3所示。通過與標準紅外吸收光譜比對,在圖3(a)中標出各吸收峰代表的分解產物。
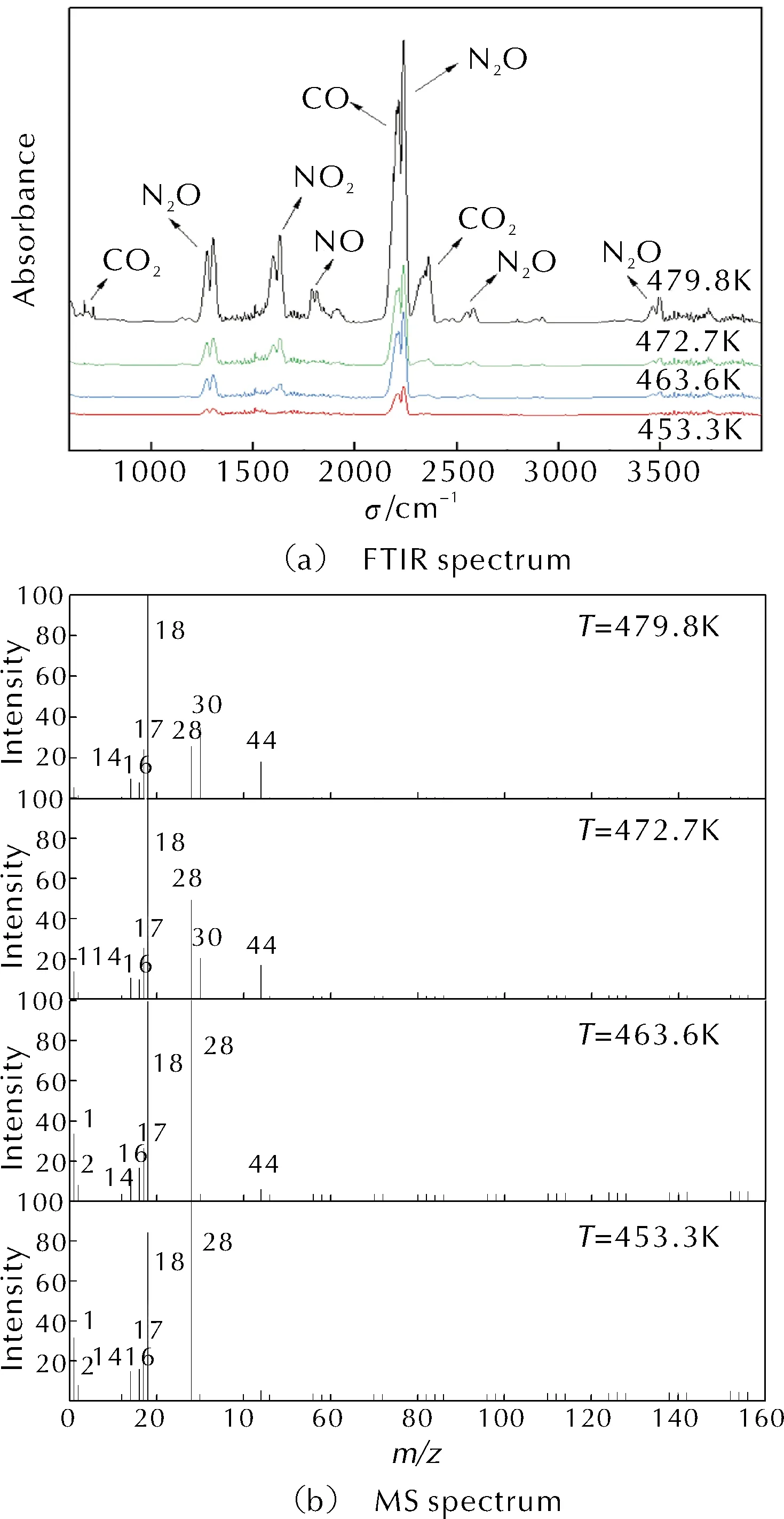
圖3 逸出氣體的紅外吸收光譜和質譜圖
由圖3可知,450~464K范圍內,HAN/PVA熱分解首先生成了H2O、H2、CO、NH3、N2O及NO2,N2O分解產生N2并將部分H2氧化成H2O;溫度逐漸升高至473K左右,H2基本被氧化成H2O,氣體產物中H2O和N2O的含量增大,NO2發生分解生成了NO,并將NH3和部分CO氧化為NO和CO2;最終當溫度升高至480K左右時,NO的含量超過了CO和N2。HAN/PVA樣品熱分解的氣相產物中,H2、CO、N2O和NO的產生可歸因于配方的氧燃比偏小。
2.2 二茂鐵類衍生物對HAN/PVA熱分解過程影響
HAN/PVA存在點火困難問題,本研究在配方中添加了質量分數1%的卡托辛、乙基二茂鐵、辛基二茂鐵或叔丁基二茂鐵作催化劑,以期改進HAN/PVA的點火燃燒性能。升溫速率為10K/min時,樣品1~樣品5的TGA-DSC曲線如圖4所示,熱分解相關的特征值見表2。
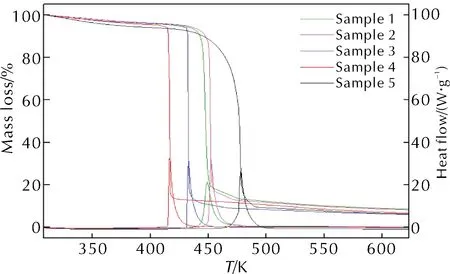
圖4 樣品1~樣品5的TGA-DSC曲線
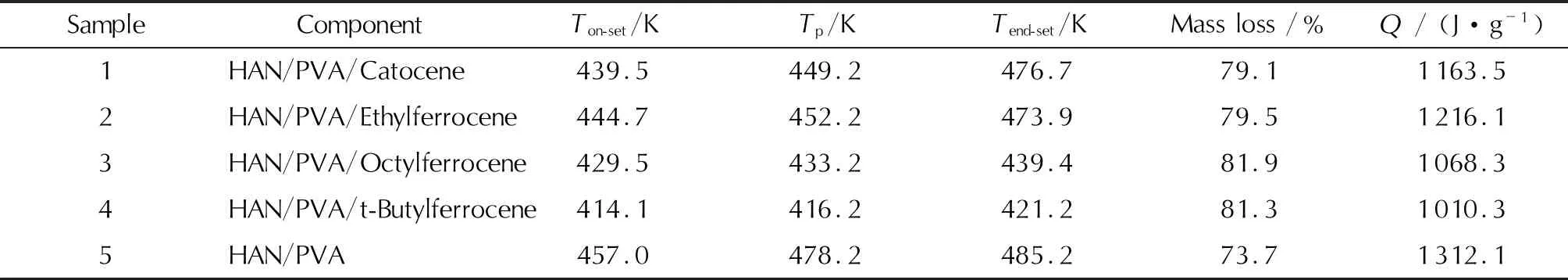
表2 二茂鐵類衍生物催化HAN/PVA熱分解特征值
注:Ton-set為起始分解溫度;Tp為峰值溫度;Tend-set為終止分解溫度。
圖4曲線表明,加入質量分數1%的二茂鐵類衍生物后,樣品1~樣品4的熱分解過程與HAN/PVA相同,熱分解的DSC曲線均僅有一個明顯的放熱分解峰。表2中數據顯示,樣品1~樣品5的起始分解溫度和分解峰值溫度從低到高依次為:樣品4<樣品3 <樣品1<樣品2<樣品5,終止分解溫度為:樣品4<樣品3<樣品2<樣品1<樣品5,熱分解持續時間為:樣品4<樣品3<樣品5<樣品2<樣品1。上述結論表明,叔丁基二茂鐵和辛基二茂鐵加快了HAN/PVA熱分解速率,反應進行地比較完全;卡托辛和乙基二茂鐵雖然使HAN/PVA的熱分解溫度提前,但是樣品熱分解產生的固相產物覆蓋在催化劑表面,可能使其反應活性降低,導致了主反應后期的熱分解速率下降,反應時間變長,反應不夠完全,殘余量略大。當升溫速率為7、15或25K/min時,5種樣品的熱分析曲線表現出相同的規律,即4種二茂鐵類衍生物對HAN/PVA熱分解的催化順序不受升溫速率的影響。
此外,從表2可以看出,樣品1~樣品4的失重率比樣品5高,且樣品的熱分解溫度大幅提前,因此二茂鐵類衍生物能夠有效促進HAN/PVA的熱分解反應。研究表明,二茂鐵類衍生物對固體推進劑的催化作用主要發生在氣相中,在凝聚相界面上也有催化作用[18]。因此,在樣品熱分析的升溫過程中,二茂鐵類衍生物首先失去茂環上的取代基,生成不穩定的二茂鐵正離子,HAN內部發生質子傳遞過程并開始分解;然后二茂鐵正離子分解成裸露的金屬離子或被氧化成粒度更小的Fe2O3,這些產物靠近樣品表面并催化HAN/PVA的熱分解過程,HAN/PVA的初始分解產物繼續分解生成了CO、CO2、N2、NO、NO2、N2O和H2O,反應放出的部分熱量加熱樣品從而進一步加快了熱分解反應進程。表2中放熱量數據顯示,4種二茂鐵類衍生物催化劑的加入均使得HAN/PVA熱分解的放熱量降低,減少量在7.3%~23.0%之間,這可能是因為催化分解反應放出的熱量積聚在氣相中,導致了系統可測得的放熱量偏低。
圖4和表2中5種配方的TGA和DSC測試結果表明,二茂鐵類衍生物使得HAN/PVA的熱分解溫度顯著降低,因此具有明顯的催化效果,其中叔丁基二茂鐵的催化效果最好,之后依次為辛基二茂鐵、卡托辛及乙基二茂鐵,使HAN/PVA的分解溫度分別降低了約42.9、27.5、17.5和12.3K。
2.3 HAN/PVA熱分解動力學參數分析
圖4中樣品4的熱分解曲線表明,加入叔丁基二茂鐵后樣品的熱分解反應瞬間完成,已經不受動力學控制,因此本研究對樣品1~樣品3和樣品5的4種配方樣品的主反應區進行動力學分析。根據式(1)將樣品在升溫速率為7、10、15和25K/min時測得的TGA曲線變換為轉化率和溫度關系曲線(α—T曲線)。
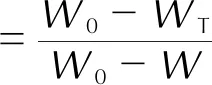
(1)
式中:α為轉化率;W0為初始質量百分數;WT為溫度T時的質量百分數;W為熱分解結束時的剩余質量百分數。
以0.05為間距,在0.3<α<0.8范圍內,求得4條α—T曲線上不同α值對應的溫度Tα,并利用等轉化率法可求出各轉化率αi下的活化能值。本研究利用Flynn-Wall-Ozawa法、Kissinger-Akahira-Sunose法和Starink法3種無模型法計算4種樣品的活化能Ea,通用式分別為式(2)、式(3)和式(4)[19-21]:
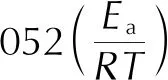
(2)
(3)
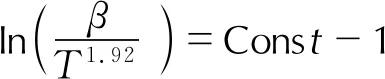
(4)
式中:β為升溫速率,K/s。
Flynn-Wall-Ozawa法擬合ln(βi)-1/Ti、Kissinger-Akahira-Sunose法擬合ln(βi/Ti2)-1/Ti、Starink法擬合ln(βi/Ti1.92)-1/Ti來確定各轉化率αi下的活化能Ei。將每種方法在不同轉化率αi下的Ei作圖,由于3種方法的Ei變化趨勢一致,故本研究僅展示Starink法求解得到的4種樣品熱分解過程αi所對應的Ei,如圖5所示。
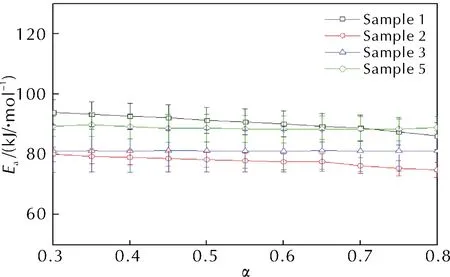
圖5 Starink法下不同轉化率對應的活化能
由圖5可知,4種配方樣品的活化能在其熱分解過程中基本保持不變,因此樣品主反應區的活化能Ea即為各個轉化率下活化能的平均值,3種方法的結果見表3。
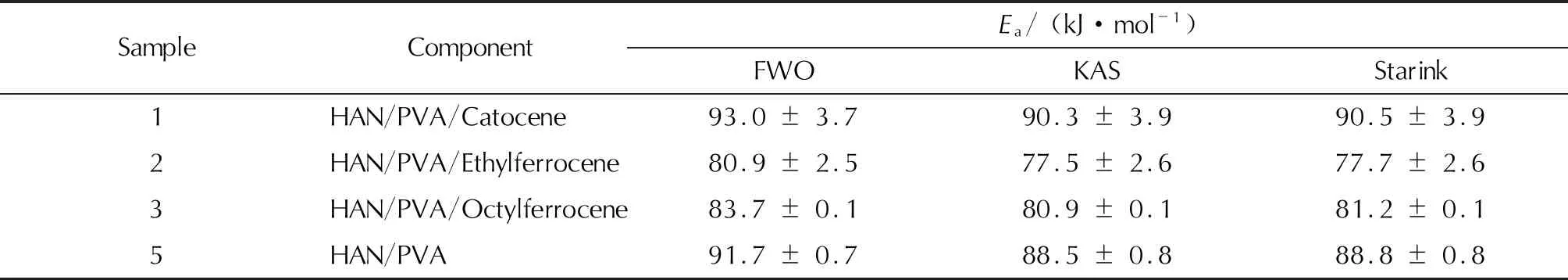
表3 不同計算方法下樣品的活化能Ea
注:Ea取轉化率在0.3~0.8之間Ei的平均數值。
對比表3中數據可知,3種方法計算得到的活化能值相差不大,由于Starink法相對于其他2種方法的準確性較高[22],本研究將Starink法求得的樣品活化能作為真值,則Flynn-Wall-Ozawa法和Kissinger-Akahira-Sunose法的相對誤差均小于5%。
HAN/PVA樣品的指前因子A可由Kissinger方程計算[22]:
(5)
式中:Tp為峰值溫度,K;f(α)為反應機理函數。
作ln(β/Tp2)-1/Tp關系圖,截距即為A。將不同升溫速率下樣品的Tpi特征值代入公式即求得樣品1~樣品3和樣品5的A分別為4.49×108(±1.72×107)、2.77×105(±9.44×103)、1.53×106(±2.27×103)和1.06×106(±1.00×104) s-1。
由表3可知,卡托辛的加入使得HAN/PVA熱分解反應活化能升高,但是圖4的TGA-DSC曲線表明其對HAN/PVA的熱分解具有明顯的催化效果。究其原因,由Arrhenius公式可知:
k=Aexp(-Ea/RT)
(6)
式中:k為反應速率常數,s-1;T為熱力學溫度,K。
樣品1~樣品3和樣品5的反應速率常數k分別為1.34×10-2、2.93×10-4、2.47×10-4和2.11×10-4s-1。
從式(6)可以看出,k受反應動力學參量Ea和A的影響,雖然HAN/PVA/卡托辛樣品的Ea和A均較大,但是其A與HAN/PVA相比提高了2個數量級,k也高出約2個數量級,即A對反應速率的影響遠超過Ea的影響,因此卡托辛的加入使HAN/PVA的反應速率常數大幅增加、熱分解速率加快,催化效果顯著。
3 結 論
(1)HAN/PVA在10K/min條件下熱分解的主反應區為457.0~485.2K,失重約73.7%,放熱量約為1312.1J/g,主要產物有CO、CO2、N2、NO、NO2、N2O和H2O。
(2)4種二茂鐵類衍生物均使得HAN/PVA的熱分解溫度降低,催化效果明顯,催化作用從強到弱依次為叔丁基二茂鐵、辛基二茂鐵、卡托辛及乙基二茂鐵,分別使HAN/PVA的熱分解溫度降低了約42.9、27.5、17.5和12.3K。
(3) 對HAN/PVA/卡托辛、HAN/PVA/乙基二茂鐵、HAN/PVA/辛基二茂鐵和HAN/PVA樣品,3種無模型法計算的活化能近似,相對誤差小于5%,熱分解反應Ea分別為90.5、77.7、81.2和88.8kJ/mol,A分別為4.49×108、2.77×105、1.53×106和1.06×106s-1。HAN/PVA/卡托辛樣品的熱分解反應速率受A的影響遠大于Ea,相應的反應速率常數大幅提高,故催化作用顯著。
