超高效液相色譜-串聯質譜法同時測定水產品中三苯甲烷類染料和氯霉素類藥物的殘留量
2020-04-10 02:15:12萬承波萬建春羅秋紅占春瑞
南昌大學學報(理科版) 2020年6期
萬承波,萬建春,羅秋紅,占春瑞*
(1.江西省產品質量監督檢測院,江西 南昌 330052;2.南昌海關技術中心,江西 南昌 330038)
三苯甲烷染料是一類工業染料,因其有較好的殺菌作用,它早期作為驅蟲劑、殺蟲劑、防腐劑在水產養殖業中使用,用于預防與治療各類水生動物的水霉病、鰓霉病和小瓜蟲病。氯霉素類藥物是類廣譜抗生素,能夠預防和治療動物各種細菌性疾病,早期被用于畜牧水產養殖[1]。三苯甲烷染料和氯霉素類藥物具有殘留時間長,三苯甲烷染料存在致癌、致畸、致突變等副作用[2],氯霉素類藥物也具有嚴重的毒副作用,它可以抑制骨髓造血機能,引起再生障礙性貧血和其他惡性血液病[3],因此世界上許多國家都禁止這兩類物質在水產養殖業的使用,我國農業農村部250號公告也將氯霉素和孔雀石綠列入《食品動物中禁止使用的藥品及其他化合物清單》中。
目前水產品中孔雀石綠(Malachite green,MG)、結晶紫(crystalviolet,CV)、及其代謝物隱性孔雀石綠(1eucom alchite green,LMG)、隱性結晶紫(1eucocrysta lviolet,LCV)、氯霉素(Chloramphenicol,CAP)、甲砜霉素(Thiamphenicol,TAP)、氟甲砜霉素(Florfenicol,FF)殘留量的檢測方法的研制受到廣泛關注,主要有免疫吸附法[4-5]、熒光光譜法[6]、高效液相色譜法[7-8]、氣相色譜串聯質譜法[9],液相色譜串聯質譜法[10-15]、液相色譜串聯高分辨質譜法[16],前處理采用乙腈或乙酸鹽緩沖液提取,固相萃取凈化富集等技術。氣相色譜串聯質譜法需經過衍生硅烷化,過程較復雜,液相色譜串聯質譜法前處理相對簡單,檢測靈敏度高。雖然目前水產品中三苯甲烷染料和氯霉素類藥物殘留量的檢測手段呈現多樣化,但兩類物質同時檢測的方法未見報道,本文建立了水產品中三苯甲烷染料和氯霉素同時檢測的方法,該方法在測定低限滿足現行法規標準的要求的同時,提高了檢測效率和降低了檢測成本。
1 實驗部分
1.1 儀器和材料
API 4000+型四極桿串聯質譜儀,美國AB SCEIX公司,配島津LC-30A超高效液相色譜儀,日本SHIMADZU公司;IKA T25高速均質機,德國IKA公司;常速離心機,上海安亭公司;KQ-600B型超聲波清洗器,昆山市超聲儀器有限公司;N-EVAPTM112型氮吹濃縮儀,美國Organomatian Associates公司;SIG 560E型號旋渦混合器,美國Scientific Industries公司;Option-Q15型超純水一體機,英國ELGA公司;固相萃取裝置(美國Supelco公司)。
孔雀石綠草酸鹽(Malachite green oxalate salt,CAS 2437-29-8),隱性孔雀石綠(leucomalachite green,CAS 129-73-7),結晶紫(Basic Violet 3,CAS 548-62-9),隱性結晶紫(Leucoc rystal Violet,CAS 603-48-5),氯霉素(Chloramphenicol,CAS 56-75-7),甲砜霉素(Thiamphenicol,CAS 15318-45-3)氟甲砜霉素(Florfenicol,CAS 76639-94-6),氘代氯霉素(Chloramphenicol-D5,CAS 202480-68-0)等標準品均購自德國Dr.Ehrenstorfer公司。孔雀石綠同位素-D5,隱性孔雀石綠同位素-D6,購自上海安譜公司。甲醇(色譜純,德國Merck公司);乙腈(色譜純,德國Merck公司);乙酸銨(色譜純,上海阿拉丁生化科技股份有限公司);乙酸(色譜純,美國TEDIA公司);中性氧化鋁固相萃取小柱(美國Supelco公司);水為超純水(電阻率18.2 MΩ·cm-1)。
1.2 儀器條件
1.2.1 三苯甲烷染料儀器條件
HPLC條件:色譜柱:Waters UPLC BEH RP C18100 mm×2.1 mm,1.7 μm;流動相A:40 mmol·L-1乙酸銨(含2%乙酸)溶液;流動相B:乙腈;梯度洗脫:0~0.5 min,50% B,0.5~1.0 min,50%~95% B,1.0~3.0 min,90% B,3.0~3.5 min,90%~50% B,3.5~5.0 min,50% B,流速:0.2 mL·min-1,進樣體積:5L,柱溫:40 ℃。
MS條件:ESI正模式掃描,多反應監測(MRM),霧化氣壓力(GS1):20 psi,輔助氣壓力(GS2):60 psi,氣簾氣壓力(CUR):40 psi,離子源溫度:600 ℃,電噴霧電壓(IS):5 000 V,各化合物去簇電壓(DP)和碰撞電壓(CE)見表1。

表1 三苯甲烷染料化合物質譜參數
1.2.2 氯霉素類藥物儀器條件
HPLC條件:色譜柱:Waters UPLC BEH RP C18100 mm×2.1 mm,1.7 μm;流動相A:水,流動相B:乙腈;梯度洗脫:0~1.0 min,30% B,1.0~2.0 min,30%~60% B,2.0~4.0 min,60%~90% B,4.0~4.1 min,90%~30% B,4.1~6.0 min,30% B,流速:0.2 mL·min-1,進樣體積:5L,柱溫:40 ℃。
MS條件:ESI負離子掃描,多反應監測(MRM),霧化氣壓力(GS1):50 psi,輔助氣壓力(GS2):50 psi,氣簾氣壓力(CUR):10 psi,離子源溫度(TEM):500 ℃,電噴霧電壓(IS):-4 500 V,各化合物去簇電壓(DP)和碰撞電壓(CE)見表2。

表2 氯霉素類藥物質譜參數
1.3 實驗方法
1.3.1 標準溶液的配制
準確稱取適量三苯甲烷染料標準品和內標物,用乙腈溶解配成1.0 mg·mL-1的標準儲備液,用棕色玻璃瓶盛放,于-18 ℃下保存。準確稱取適量氯霉素類標準品和內標物,用甲醇溶解配成100 μg·mL-1的標準儲備液,用棕色玻璃瓶盛放,于-18 ℃下保存。分別移取適量的氯霉素類藥物和三苯甲烷染料標準儲備液,用乙腈逐級稀釋成10、50、100 ng·mL-1的標準中間液。分別移取內標物儲備液用,乙腈逐級稀釋成100 ng·mL-1內標物中間液。
1.3.2 定容液和流動相的配制
40 mmol·L-1乙酸銨溶液:準確稱取3.15 g乙酸銨,用純水溶解并定容至1 000 mL;乙腈-40 mmol·L-1乙酸銨溶液(1+1,v:v):準確量取50 mL乙腈,加入50 mL 40 mmol·L-1乙酸銨溶液,混合均勻;40 mmol·L-1乙酸銨(含2%乙酸)溶液:向998 mL 40 mmol·L-1乙酸銨溶液中加入2 mL乙酸,混合均勻。
1.3.3 樣品前處理
稱取試樣5.0 g,置于50 mL聚丙烯離心管內,加入11 mL乙腈,高速均漿提取30 s,在4 500 r·min-1下離心5 min,上層清液轉入25 mL比色管中,殘渣中再加入11 mL乙腈,渦旋混合30 s,超聲提取5 min,在4 500 r·min-1下離心5 min,合并上層清至25 mL比色管中,用乙腈定容,混合均勻后待凈化。取5 mL提取液倒入經5 mL乙腈活化平衡的中性氧化鋁小柱內,再用5 mL乙腈洗脫,收集全部流出液,洗脫液在45 ℃以下水浴,氮吹至干,殘渣用2 mL定容液定容,超聲溶解后過0.22 μm濾膜,供UHPLC-MS/MS測定。
2 結果與討論
2.1 質譜條件的優化
蠕動泵將標準溶液泵入質譜儀,三苯甲烷染料在ESI正模式下有最佳的響應值,質譜全掃描獲得MG和CV及其代謝物的分子峰[M+H]+,加上碰撞電壓時,MG、CV及其代謝物,都會產生失去一個甲基和一個二甲基苯胺碎片離子。氯霉素類藥物在負模式下掃描響應值較高,質譜全掃獲得其分子峰[M-H]-,加上碰撞電壓時,其結構中2-二氯乙酰胺基都易失去或發生結構重組。歐盟2002/675 EC指令規定,質譜確證方法需要4個確證點數,每個待物選擇兩對離子,作為定性離子,同位素內標物選擇一對定量參比離子。確定掃描離子對后,聯接液相進樣,對質譜離子源參數進行優化,自動優化獲得最佳電噴霧電壓(IS)、霧化氣壓力(GS1)、輔助氣壓力(GS2)、氣簾氣壓力(CUR)、離子源溫度(TEM)。
2.2 色譜條件的選擇
三苯甲烷染料和氯霉素類藥物為中等極性到弱極性化合物,在反相色譜中均可保留,目前多數文獻和標準方法均采用反相色譜進行分離[15-17]。三苯甲烷類藥物正模式掃描,在流動相中加入一定揮發性酸能促進化合物的電離,可以提高靈敏度,而氯霉素類藥物在負模式掃描,加入酸會抑制化合物的電離,導致響應值下降,兩類化合物在同一色譜條件下進行分析,靈敏度比較難兼顧,因此本方法確定兩類采用不同儀器條件進行分析檢測。
實驗比較了不同的色譜柱,發現Waters UPLC BEH RP C18柱(100 mm×2.1 mm,1.7 μm)相比Waters UPLC HSS T3柱(50 mm×1.0 mm,1.8 μm)分離效果較好,化合物響應值更高,因此確定Waters UPLC BEH RP C18柱為本方法分析用色譜柱。
2.3 提取和凈化條件的選擇和優化
多數文獻報道采用乙酸乙酯、乙腈等有機溶液提取水產品中氯霉素類藥物,三苯甲烷染料及其代謝物[8,18-19],本方法比較了乙腈和乙酸乙酯不同的提取效率,實驗發現乙腈提取整體效率要做優于乙酸乙酯,尤其是LMG,乙酸乙酯提取回收率都在20%以下,而乙腈的提取回收率均在85%以上,TAP和MG用乙酸乙酯提取的回收率在50%以下,乙腈的提取回收率除鰻魚的回收率略低于70%,在小龍蝦和鯉魚提取回收率都超過了80%,可能是因為鰻魚中含油脂較多的原因,由于部分藥物的極性相對較強,乙酸乙酯提取效率不高,乙酸乙酯提取的油脂較多,基質效應可能也影響了提取回收率,因此本方法確定乙腈作為提取溶劑。
水產品中三苯甲烷染料和氯霉素類藥物殘留量較低,樣品在提取后需進行進一步純化富集。液液萃取操作繁瑣且有機溶劑消耗量大,冷凍除油凈化[19],固相萃取凈化HLB柱[20-21]等凈化方式均有相關報道,但兩類藥物同時純化的處理方式未見報道。三苯甲烷染料和氯霉素類藥物的極性跨度大,且三苯甲烷染料在水溶液中為陽離子,采用反相或離子交換作用機理的固相萃取方式,可能難以兼顧兩類藥物,本方法采用正相純化的方式,以除去提取液共萃取油脂等非極性雜質。中性氧化鋁柱是一種正相保留機理的固相萃取柱,能夠吸附極性化合物,并去除油脂。本方法采用中性氧化鋁柱凈化,能夠去除共萃取物中的油脂,同時對目標物不會產生死吸附,對比了不同品牌的中性氧化鋁小柱,除品牌C的穩定性略差外,品牌間純化效果的差異并不明顯。同時考查了洗脫體積對回收率的影響,5 mL乙腈洗脫基本能將目標物全部洗脫下來,再增加5 mL乙腈洗脫未檢出目標物,且濃縮后殘渣有一些增加,考慮后續濃縮時雜質干擾的影響,洗脫體積確定為5 mL。
2.4 特異性
本方法用空白鰻魚、小龍蝦、鯉魚樣品與其對照加標樣品,進行全過程實驗,實驗結果表明兩類藥物均具有良好的特異性,在目標物出峰處無干擾峰。
2.5 基質效應
基質效應是影響質譜法定量結果重要因素,本方法考查了基質效應對定量結果的影響,用空白基質溶液配制標準曲線與試劑配制標準曲線比較,通過計算基質標準曲線與試劑過原點標準曲線斜率比值的關系,確定基質效應。實驗結果表明鰻魚、小龍蝦和鯉魚基質中各化合物存在不同的基質效應作用,而且基質作用的強弱有所差異,三苯甲烷染料基質效應非常大,氯霉素類藥物也存在一定的基質效應。在定量時采用同位素內標可以消除基質效應,但本方法所測定的7種化合物只有3種化合物有同位素內標,為有效消除基質效應,本方法確定采用基質標準曲線定量,孔雀石綠、隱性孔雀石綠、氯霉素采用同位素內標定量,其它4種化合物采用基質外標法定量。
2.6 測定低限和線性范圍
用空白鰻魚、小龍蝦、鯉魚加標實驗,通過計算目標物的信噪比,以信噪比大于10的濃度確定定量限,三苯甲烷染料定量限為0.5 μg·kg-1,氯霉素定量限為0.1 μg·kg-1,甲砜霉素和氟甲砜霉素定量限為1.0 μg·kg-1。考查了基質標準曲線的線性范圍,三苯甲烷染料在0.25~5.0 μg·kg-1,氯霉素在0.05~2.0 μg·kg-1,甲砜霉素和氟甲砜霉素在0.5~20.0 μg·kg-1濃度范圍內呈線性關系,線性相關系數均在0.99以上。
2.7 準確度和精密度
用空白鰻魚、小龍蝦、鯉魚樣品加標方式進行準確度和精密度實驗,實驗分別在1、2和4倍定量限濃度水平進行實驗,加標回收率在61.5~119.0%之間,RSD值在0.76~14.46%之間。

表3 7種藥物在鯉魚基質中的定量限、回歸方程、相關系數(r)、回收率及相對標準偏差(n=6)
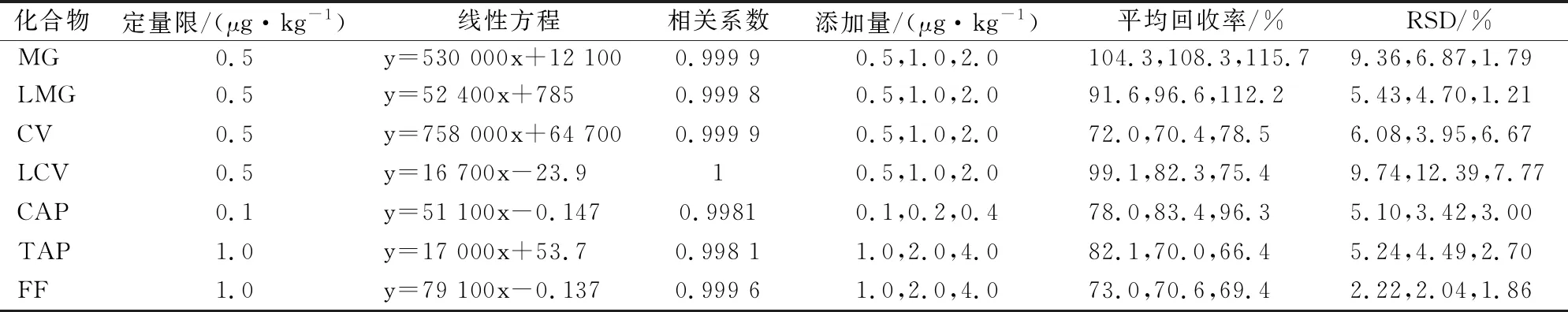
表4 7種藥物在鯉魚基質中的定量限、回歸方程、相關系數(r)、回收率及相對標準偏差(n=6)
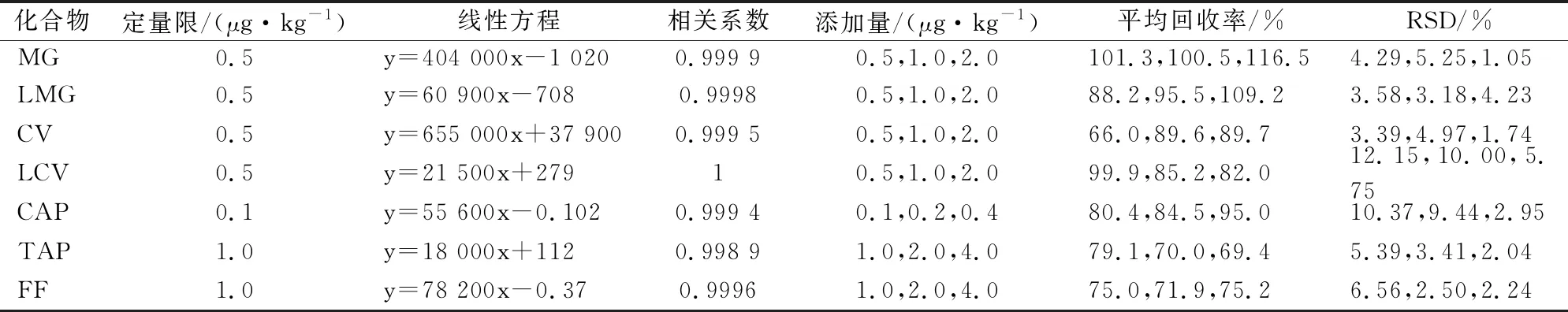
表5 7種藥物在鯉魚基質中的定量限、回歸方程、相關系數(r)、回收率及相對標準偏差(n=6)
3 實際樣品檢測
本方法建立的水產品中三苯甲烷染料和氯霉素類藥物殘留量同時檢測的方法,操作簡單、特異性強、靈敏度高。實際檢測100多批次樣品,其中檢出1批次鰻魚氟甲砜霉素殘留陽性,實際運用證明本方法準確可靠,該方法可適用于鰻魚、小龍蝦、鯉魚等產品的三苯甲烷染料和氯霉素類藥物殘量的同時測定。
