奧拉西坦膠囊中主要未知雜質的鑒定及其含量測定
2020-04-08 01:21:03張菁孫婷趙昌夢姜建國
中國藥房 2020年6期
張菁 孫婷 趙昌夢 姜建國

ABSTRACT? ?OBJECTIVE: To identify the main unknow impurity of Oxiracetam capsule and determine its content, so as to improve the standard of quality control. METHODS: Two-dimensional UPLC-IT-TOF-MS was adopted to qualitatively analyze the unknown impurity. One-dimensional liquid chromatogram analysis was performed on ST PAK C18 ES column with mobile phase consisted of 0.02 mol/L sodium dihydrogen phosphate solution at the flow rate of 0.5 mL/min. The column temperature was set at 30 ℃, sample size was 20 μL. The detection wavelength was set at 210 nm. Two-dimensional liquid chromatogram analysis was performed on Techmate C18-STⅡ column with mobile phase consisted of 0.02 mol/L ammonium acetate solution at the flow rate of 0.5 mL/min. The column temperature was 30 ℃. Mass spectrometry was adopted (electropray ionization source, MS+ and MS- mode data acquisition). After the target impurity was located by one-dimensional liquid chromatography, it was transferred to two-dimensional liquid chromatography-mass spectrometry system for qualitative analysis. The unknown impurity structure was inferred by means of molecular formula prediction module “Accurate Mass Calculator”in LCMS Solution, and the refined impurity products by preparation and purification were standardized and confirmed. The impurity content was determined by HPLC (with the same condition of one-dimensional liquid chromatography for qualitative analysis). RESULTS: The main unknown impurity in Oxiracetam capsules is oxiracetam acid. The content of the refined product was 99.5% after preparation and purification. The contents of oxiracetam acid in 9 batches of Oxiracetam capsules were 0.05%-0.14%. CONCLUSIONS: The established two-dementional UPLC-IT-TOF-MS method can accurately locate the peak position of the impurity oxiracetam acid, and analyze its structure, while the corresponding content determination method can better separate the impurity from the main drug and other components, with good sensitivity, precision, repeatability, stability and accuracy. The quality of the finished product of Oxiracetam capsules can be well controlled by using above method.
KEYWORDS? ?Two-dementional UPLC-IT-TOF-MS; HPLC;Oxiracetam capsule;Oxiracetam acid; Impurity; Identification; Content determination
奧拉西坦為吡拉西坦的類似物,其化學名為4-羥基-2-氧代-1-吡咯烷乙酰胺(化學結構式見圖1),臨床上主要用于改善老年性癡呆和記憶障礙患者的記憶功能[1],其作用機制是通過促進磷酰膽堿和磷酰乙醇胺的合成,提高大腦中腺苷三磷酸(ATP)/腺苷二磷酸(ADP)的比值,增加大腦中蛋白質和核酸的合成[2]。按照2015年版《中國藥典》(四部)相關指導原則[3]開展的穩定性試驗結果顯示,奧拉西坦膠囊在貯存過程中雜質含量有逐漸增加的趨勢。奧拉西坦原料藥現行注冊質量標準對其有關物質采用高效液相色譜(HPLC)自身對照法進行測定[4-5],但無法對有關物質的含量進行準確控制;而奧拉西坦膠囊質量標準則未對有關物質進行控制[6]。基于此,本課題組參考相關文獻[7-8],建立了超高效二維液相色譜-離子阱-飛行時間質譜法,對奧拉西坦膠囊中的主要未知雜質進行定性分析,確定雜質的結構,并對該雜質進行含量測定,為提高奧拉西坦膠囊質量控制標準、確保其臨床使用的有效性和安全性提供技術支持。
1 材料
1.1 儀器
2D-LC-IT-TOF/MS型超高效二維液相色譜-離子阱-飛行時間質譜系統、SPD-M20A型二極管陣列檢測器(用于定性分析)、LC-20AD型液相色譜儀(用于含量測定)均購自日本Shimadzu公司;Mercury-400型核磁共振(NMR)儀(美國Varian公司);CP225D型十萬分之一電子天平(德國Sartorius公司)。
1.2 藥品與試劑
奧拉西坦膠囊(廠家A,批號:20180701、20180702、20180921;廠家B,批號:264180627、264180268、264180269;廠家C,批號:180801、180802、180803;規格均為0.4 g);奧拉西坦原料藥[石藥集團歐意藥業(石家莊)有限公司,批號:264100627,純度:99.3%];醋酸銨、冰醋酸、二氯甲烷、甲醇均為色譜純,氫氧化鈉、濃鹽酸、磷酸二氫鈉均為分析純,水為超純水。
2 方法與結果
2.1 奧拉西坦膠囊中主要雜質的定性分析
2.1.1 色譜條件 (1)一維液相色譜條件:色譜柱為ST PAK C18 ES柱(250 mm×4.6 mm,5 μm),流動相為0.02 mol/L磷酸二氫鈉溶液,流速為0.5 mL/min,柱溫為30 ℃,檢測波長為210 nm,進樣量為20 μL。(2)二維液相色譜條件和質譜條件:色譜柱為Techmate C18-STⅡ柱(150 mm×4.6 mm,5 μm),流動相為0.02 mol/L醋酸銨溶液(用冰醋酸調節pH為4.8),流速為0.5 mL/min,柱溫為30 ℃;離子源為電噴霧離子源(ESI),離子源溫度為500 ℃,接口電壓為4 500 V(正離子模式)、-4 500 V(負離子模式),掃描方式為一級質譜全掃描(正、負離子模式),去簇電壓為55 V(正離子模式)、-45 V(負離子模式)。
2.1.2 溶液的制備 (1)供試品溶液:精密稱取奧拉西坦膠囊內容物適量,加0.02 mol/L磷酸二氫鈉溶液溶解并稀釋制成每1 mL中含奧拉西坦1 mg的溶液,即得。(2)自身對照溶液:精密量取上述供試品溶液0.5 mL,置于100 mL量瓶中,加0.02 mol/L磷酸二氫鈉溶液稀釋至刻度,搖勻,即得。
2.1.3 主要未知雜質定性分析 精密量取“2.1.2”項下自身對照溶液和供試品溶液各20 μL,按“2.1.1”項下條件注入液相色譜儀,經一維液相色譜條件分離確定主要未知雜質的保留時間為6.607 min,詳見圖2。將該未知雜質(保留時間為6.607 min的部分)切換到LOOP環中,進行二維脫鹽分析,記錄總離子流圖和未知雜質質譜圖,詳見圖3、圖4。結果顯示,在奧拉西坦一維液相色譜圖中保留時間為6.607 min的未知雜質峰,其分子離子峰分別為m/z 159.9[M+H]+和m/z 158.1[M-H]-,分子量為159。采用2D-LC-IT-TOF/MS系統的LCMS Solution Ver 3.70 色譜工作站中的分子式預測模塊“Accurate Mass Calculator”,結合化合物氫碳比、氮規則、質譜信息以及奧拉西坦雜質合成路線[9],預測該雜質的可能分子式為C6H9NO4,初步推斷該雜質為奧拉西坦酸(化學結構式見圖1)。
2.1.4 破壞試驗 取奧拉西坦原料藥適量,經0.2 mol/L鹽酸溶液或0.2 mol/L氫氧化鈉溶液分別進行酸、堿破壞后,按“2.1.2(1)”項下方法制成兩種破壞樣品供試品溶液后,再按“2.1.1(1)”項下色譜條件進樣測定(圖略)。結果顯示,在一維色譜圖保留時間約6.6 min處的色譜峰明顯增大,經二極管陣列檢測器檢測后顯示,其最大吸收波長為197 nm;然后按“2.1.1(2)”項下條件檢測后顯示,該雜質分子量為159,推測其為奧拉西坦在酸、堿作用下水解生成的奧拉西坦酸。
2.2 雜質奧拉西坦酸的制備純化、標化及結構確證
2.2.1 雜質奧拉西坦酸的制備純化 參考文獻方法[9]進行制備與純化。取奧拉西坦原料藥20 g,置于500 mL回流瓶中,加入0.5 mol/L氫氧化鈉溶液100 mL,攪拌使溶解,加熱保溫回流反應5 h后停止;冰浴降溫至0 ℃后,用1 mol/L鹽酸溶液調節pH為4,在50 ℃條件下蒸發至近干后,采用Sep-Pak?Silica固相萃取小柱層析,以二氯甲烷和甲醇梯度洗脫;收集洗脫液,在30 ℃條件下蒸發至干,得黃色油狀物。取該油狀物,加入90%甲醇50 mL后,在60 ℃條件下加熱使溶解,趁熱過濾;濾液于0 ℃條件下放置過夜,析出結晶后濾過;結晶以甲醇清洗后,在60 ℃條件下干燥,得白色固體,即得雜質奧拉西坦酸精制品。
2.2.2 奧拉西坦酸的標化及結構確證 取“2.2.1”項下制備純化所得的奧拉西坦酸精制品適量,加0.02 mol/L磷酸二氫鈉溶液溶解制成每1 mL中含奧拉西坦酸1 mg的溶液,再按“2.1.1(1)”項下色譜條件進樣測定。采用峰面積歸一化計算,結果所制奧拉西坦酸的含量為99.5%(n=6)。經1H-NMR(400 MHz,D2O)分析,結果顯示,δ:4.40(m,1H),4.14(d,2H,J=17.6 Hz),3.92(dd,1H,J=17.6 Hz,6.5 Hz),3.42(dd,1H,J=10.4 Hz,1.9 Hz),2.87(dd,1H,J=17.2 Hz,6.4 Hz),2.28(dd,1H,J=17.2 Hz,2.0 Hz)。
2.3 奧拉西坦膠囊中奧拉西坦酸的含量測定
采用HPLC法進行含量測定。
2.3.1 色譜條件 采用“2.1.1(1)”項下色譜條件。
2.3.2 溶液的制備 (1)供試品溶液:按“2.1.2(1)”項下方法配制,即得。(2)自身對照溶液:取上述供試品溶液按“2.1.2(2)”項下方法配制,即得。(3)雜質對照品溶液:精密稱取“2.2.1”項下奧拉西坦酸精制品25 mg,置于25 mL量瓶中,加0.02 mol/L磷酸二氫鈉溶液溶解并稀釋至刻度,搖勻,作為貯備液;臨用前取該貯備液0.5 mL,置于100 mL量瓶中,加0.02 mol/L磷酸二氫鈉溶液溶解并稀釋至刻度,搖勻,即得。(4)空白輔料溶液;取膠囊輔料(本課題組根據各廠家處方自行配制)按照處方比例混合,精密稱取適量,加0.02 mol/L磷酸二氫鈉溶液溶解制成空白輔料溶液。
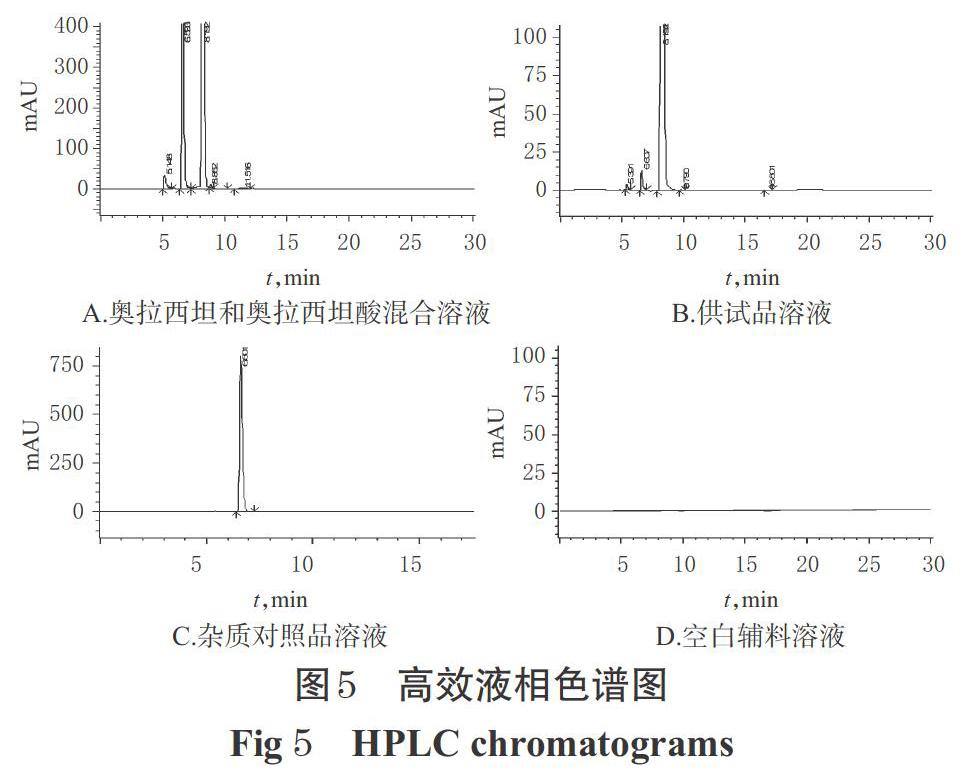
2.3.3 系統適用性試驗 稱取“2.2.1”項下奧拉西坦酸精制品適量,加入“2.3.2(2)”項下自身對照溶液適量,加0.02 mol/L磷酸二氫鈉溶液稀釋制成含奧拉西坦1 μg/mL、奧拉西坦酸5 μg/mL的混合溶液,再按“2.1.1(1)”項下色譜條件進樣測定;另取“2.3.2(1)(3)(4)”項下供試品溶液、雜質對照品溶液、空白輔料溶液同法進樣測定,記錄色譜圖,詳見圖5。結果顯示,奧拉西坦和奧拉西坦酸分離度大于4.6,理論板數按奧拉西坦峰計不低于5 000,空白輔料無干擾。
2.3.4 檢出限和定量限考察 取“2.3.2(3)”項下雜質對照品溶液,以0.02 mol/L磷酸二氫鈉溶液為溶劑逐級稀釋制得系列溶液,按“2.1.1(1)”項下色譜條件進樣測定。結果,當信噪比分別為3 ∶ 1、10 ∶ 1時,得奧拉西坦酸的檢出限為0.02 ng、定量限為0.1 ng。
2.3.5 線性關系考察 取“2.3.2(2)(3)”項下自身對照溶液和雜質對照品溶液,按“2.1.1(1)”項下色譜條件分別進樣5、10、20、40、80 μL,記錄峰面積。分別以奧拉西坦和奧拉西坦酸的進樣量(X,ng)為橫坐標、相應峰面積(Y)為縱坐標繪制標準曲線,得兩者回歸方程分別為:奧拉西坦Y=2 007.2X-2 647.2(r=1.000),奧拉西坦酸Y=2 821.0X-15 783.0(r=0.999 6)。結果表明,奧拉西坦和奧拉西坦酸進樣量均在25~400 ng范圍內與其峰面積呈良好的線性關系。
2.3.6 精密度試驗 精密量取“2.3.3”項下奧拉西坦和奧拉西坦酸混合溶液 20 μL,按“2.1.1(1)”項下色譜條件連續進樣測定6次,記錄峰面積。結果,奧拉西坦和奧拉西坦酸峰面積的RSD分別為0.6%、0.9%(n=6),表明儀器精密度良好。
2.3.7 重復性試驗 稱取奧拉西坦膠囊內容物(批號:180802)適量,共6份,按“2.3.2(1)”項下方法制備供試品溶液,同時取“2.3.2(3)”項下雜質對照品溶液,分別按“2.1.1(1)”項下色譜條件進樣測定,記錄峰面積,按外標法計算奧拉西坦酸平均含量為0.05%(RSD=0.67%,n=6),表明本方法重復性良好。
2.3.8 加樣回收率試驗 稱取奧拉西坦膠囊內容物(批號:180802)適量,共9份,精密稱定,分別置于25 mL量瓶中,精密加入50 μg/mL雜質對照品溶液[按“2.3.2(3)”項下方法重新配制]0.8、1.0、1.2 mL,每個質量濃度平行3份。加樣后,按“2.3.2(1)”項下方法制備供試品溶液,再按“2.1.1(1)”項下色譜條件進樣測定,記錄峰面積,計算加樣回收率。結果,奧拉西坦酸平均加樣回收率為99.25%(RSD=1.6%,n=9),詳見表1。
2.3.9 穩定性試驗 取同一份供試品溶液(批號:180802),分別于室溫下放置0、2、4、6、8 h時,按“2.1.1(1)”項下色譜條件進樣測定,記錄峰面積。結果,奧拉西坦酸峰面積的RSD為0.96%(n=5),表明供試品溶液在室溫下放置8 h內基本穩定。
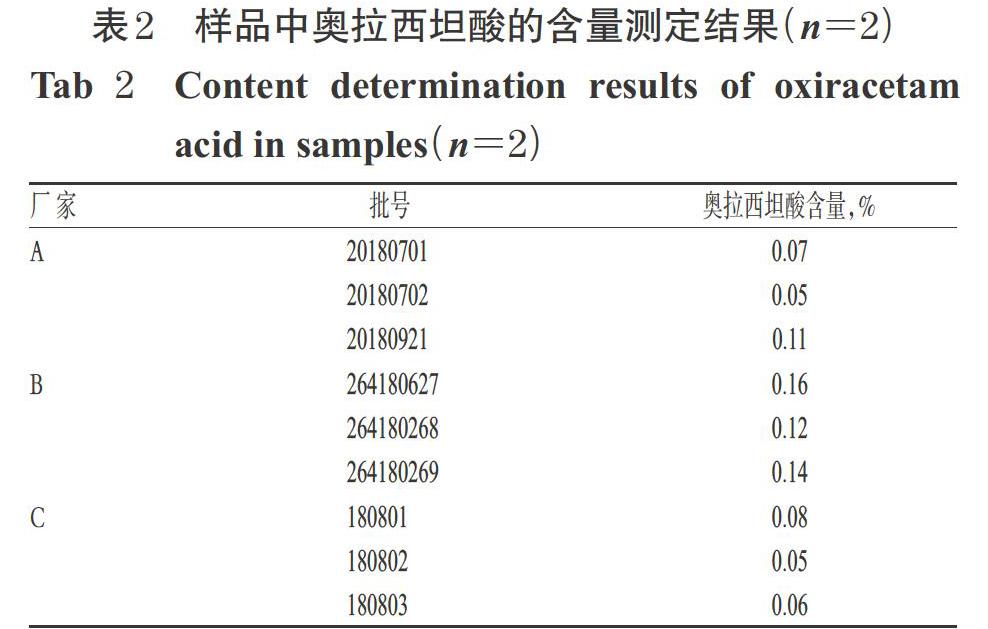
2.3.10 不同廠家9批樣品中奧拉西坦酸的含量測定取3個廠家共9批奧拉西坦膠囊樣品內容物適量,按“2.3.2(1)”項下方法制備供試品溶液,同時取“2.3.2(3)”項下雜質對照品溶液,分別按“2.1.1(1)”項下色譜條件進樣測定,記錄峰面積,采用外標法計算奧拉西坦酸含量(按膠囊規格0.4 g計算):雜質含量=(供試品溶液奧拉西坦酸峰面積×雜質對照品溶液濃度×樣品平均裝量)/(雜質對照品溶液峰面積×供試品溶液濃度×樣品規格)×100%。每批樣品平行2份測定,取平均值。結果,9批樣品中均檢出奧拉西坦酸,其測定結果詳見表2。
3 討論
3.1 色譜分析色譜柱的選擇
現行奧拉西坦膠囊標準中無有關物質測定項,而其原料藥注冊質量標準中有關物質測定采用色譜柱為C8柱或氰基柱。本課題組在相同的色譜條件下,比較了C8柱、氰基柱和C18柱的保留時間和分離效果。結果顯示,因奧拉西坦的極性大、分子小,故在C8柱上保留時間短、分離效果欠佳,主峰無法與破壞試驗中產生的雜質峰較好地分離;采用氰基柱分離時,由酸、堿破壞試驗所獲得的雜質色譜峰較少,且主峰的純度不高;采用ST PAK C18 ES色譜柱進行分析時,因該色譜柱進行了新型聚合物包被,不僅使奧拉西坦有適當的保留時間,而且對雜質的分離效果也好,故選擇其作為一維色譜分析的色譜柱。
3.2 色譜分析流動相的選擇
在確定了色譜柱的基礎上,筆者考慮到需準確定位雜質的出峰時間,故重點對一維色譜流動相進行篩選。先后選用甲醇-水、乙腈-水、水、磷酸鹽緩沖液等系統,并適當調節流動相比例后進行了一系列考察。結果顯示,流動相中如果含有機溶劑,則柱效低、保留時間短,主峰與相鄰峰不能得到較好分離;采用水為流動相則會出現較大的溶劑峰和倒峰;而采用磷酸鹽緩沖液系統可避免出現以上情況。通過試驗篩選后發現,以0.02 mol/L磷酸二氫鈉緩沖鹽為流動相時,主峰保留時間適宜,與各雜質峰的分離度良好,且峰寬較小,便于對目標雜質成分進行峰切割后轉入二維色譜分析。
3.3 奧拉西坦雜質的產生
奧拉西坦含酰胺鍵,合成中在無水碳酸鈉作用下易水解成羧酸,生成奧拉西坦酸(即4-羥基-2-氧代-1-吡咯烷乙酸)[10]。奧拉西坦酸為合成過程中的主要副產物,以及在酸性、堿性條件下的主要降解產物,可進一步與甲醇和乙醇發生酯化反應,生成4-羥基-2-氧代-1-吡咯烷乙酸甲酯和4-羥基-2-氧代-1-吡咯烷乙酸乙酯,所以控制奧拉西坦酸的含量顯得尤為重要。
綜上所述,奧拉西坦酸為奧拉西坦膠囊中的主要雜質;制備純化后獲得的奧拉西坦酸精制品含量達到99.5%,可以滿足作為對照品的要求;所建立的超高效二維液相色譜-離子阱-飛行時間質譜法能準確定位雜質奧拉西坦酸的出峰位置并對其進行結構分析,且相應的含量測定方法能較好地分離雜質與主藥及其他成分,靈敏度、精密度、重復性、穩定性、準確度均良好,符合2015年版《中國藥典》要求[3]。因此,采用單一雜質外標法測定奧拉西坦酸的含量,并結合自身對照法測定其他雜質,可較好地跟蹤測定生產制備過程中的中間體、控制奧拉西坦膠囊的成品質量。
參考文獻
[ 1 ] 張倩倩.奧拉西坦治療卒中后認知功能障礙的有效性及安全性探析[J].健康之路,2016,15(5):119-120.
[ 2 ] 金磊,李博,葉雷,等.奧拉西坦的臨床前藥理學研究[J].中國臨床藥理學與治療學,2011,16(3):354-360.
[ 3 ] 國家藥典委員會.中華人民共和國藥典:四部[S]. 2015年版.北京:中國醫藥科技出版社,2015:354-356、374-378.
[ 4 ] 國家食品藥品監督管理局.WS-1005(X-750)-2002 奧拉西坦:試行[S].2003-01-28.
[ 5 ] 國家食品藥品監督管理局. YBH06612003 奧拉西坦:試行[S]. 2003-09-27.
[ 6 ] 國家食品藥品監督管理局. YBH06622003 奧拉西坦膠囊:試行[S].2003-09-27.
[ 7 ] 沈保家,秦昆明,劉啟迪,等.二維色譜技術及其在中藥領域中的應用[J].中國科學(化學),2013,43(11):1480- 1489.
[ 8 ] 李郭帥,馬陽,耿婷,等.超高效二維液相色譜法測定復方南星止痛膏中雙酯型烏頭生物堿的含量[J].藥物分析雜志,2019,39(2):249-256.
[ 9 ] 張毅,陳瑞,湯磊.兩個奧拉西坦雜質的合成[J].化學研究與應用,2018,30(4):602-604.
[10] 吳福鴻,李兆林,楊亞軍.奧拉西坦的合成工藝改進[J].石油化工應用,2018,37(7):117-119.
(收稿日期:2019-08-01 修回日期:2019-02-12)
(編輯:段思怡)
