加壓毛細管電色譜測定甘蔗中三種多酚類化合物
2020-04-01 10:44:54,*
食品工業科技 2020年4期
關鍵詞:檢測
,*
(1.廣西科技大學生物與化學工程學院,廣西柳州 545006;2.廣西科技大學醫學院,廣西柳州 545005)
多酚類化合物普遍存在于植物界,是植物中豐富的次級代謝產物[1-2],具有很高的抗氧化能力[3]。有研究表明,多酚類化合物還有許多生物特性,如抗炎、抗過敏、抗菌、保護心臟等[4-6]。由于多酚化合物具有較高的利用價值,應用廣泛,因此,各類植物、食品中多酚化合物的分離、檢測及其質量控制等方面引起了眾多研究者的關注[7-9]。
甘蔗廣泛分布于溫帶和熱帶地區,其含糖量高,主要用作生產糖和酒精[10]。甘蔗在加工過程中會產生大量的副產物如甘蔗渣。研究表明,甘蔗是多酚含量較高的植物,在蔗葉、蔗梢、蔗莖中均含有大量的高分子多酚類化合物,其多酚含量均在為500 mg/kg以上[11-14]。多酚類化合物可與多酚氧化酶作用引起酶促褐變反應,使蔗糖顏色加深[15-16]。為了提高對甘蔗渣的充分利用率,并對蔗糖進行質量控制,因此有必要對多酚類化合物的提取、分離及檢測進行進一步研究。
甘蔗中可檢測多酚類化合物含量較低,干擾成分較多且基底復雜,目前,對于甘蔗中多酚分離檢測的研究相對較少,且不充分。目前常用的多酚的分析方法為高效液相色譜法[17-18]和毛細管電泳法[19],但高效液相色譜法多采用梯度洗脫且分析時間較長,毛細管電泳法分析快速卻常出現干柱和氣泡問題,影響柱效。加壓毛細管電色譜(pCEC)是 21 世紀初發展起來的一種新型分離技術,將液相色譜與毛細管電泳結合,實現以壓力流和電滲流作為雙重驅動力,解決了高效液相色譜法分析時間長和毛細管電泳法的干柱和氣泡問題,達到更加快速、高效的分離檢測目的[20-21]。目前加壓毛細管電色譜在藥品、食品、環境安全等方面已得到廣泛應用[22-24]。本文利用加壓毛細管電色譜,對影響分離檢測的色譜條件流動相比例、緩沖鹽種類、濃度及pH、離子對試劑濃度、分離電壓、流速等進行優化,建立了分離檢測甘蔗中綠原酸、阿魏酸、表兒茶素的方法。
1 材料與方法
1.1 材料與儀器
甘蔗 廣西柳州;綠原酸、阿魏酸、沒食子酸標準品 上海安譜實驗科技股份有限公司;甲醇 色譜純,德國Merck 公司;庚烷磺酸鈉 色譜純,天津市化學試劑研究所;磷酸二氫鈉、乙酸鈉、乙酸銨、氫氧化鈉、鹽酸、乙酸乙酯 分析純,廣東汕頭市西隴化工廠;實驗用水 均為超純水。
TriSepTM-2100 加壓毛細管電色譜 上海通微分析技術有限公司;UV-2102PC型紫外-可見分光光度計 上海尤尼柯儀器有限公司;RE-5203旋轉蒸發器 上海亞榮生化儀器廠。
1.2 實驗方法
1.2.1 標準溶液的配制 精密稱取綠原酸、阿魏酸、表兒茶素標準品各25 mg,分別置于25 mL棕色容量瓶,用甲醇溶解并定容,配制成1 mg/mL的標準溶液。用甲醇按梯度將其分別稀釋為20、40、80、160、320 μg/mL的標準溶液。所有標準溶液均在4 ℃下避光保存。
1.2.2 混合標準溶液的配制 分別精密吸取1 mg/mL的綠原酸、阿魏酸、表兒茶素標準溶液各80 μL,用甲醇稀釋至1 mL,作為混合標準溶液備用。
1.2.3 多酚的提取 參考Fang等[25]的提取方法,將所購甘蔗去皮后榨去蔗汁,取甘蔗渣適量干燥后,研磨成粉。準確稱取甘蔗渣粉末10.00 g,加入1 mol/L NaOH溶液150 mL,室溫下磁力攪拌6 h后,用6 mol/L HCl溶液調節pH至2.0,移至分液漏斗中,用乙酸乙酯萃取三次,將萃取液放至旋轉蒸發儀中,40 ℃旋轉蒸干,得到紅棕色膏狀物(為萃取后除脂及多糖之后,富含多酚類化合物的混合物),放至冰箱4 ℃保存。取適量上述棕紅色膏狀物,用甲醇溶解,通過0.20 μm的針式微孔濾膜過濾,冰箱4 ℃保存備用。
1.2.4 pCEC色譜條件 色譜柱:毛細管填充柱(EP-100-20/45-3-C18,內徑100 μm,總長度45 cm,有效長度20cm,填料為內徑3 μm OSD);流動相:15.0 mmol/L NaH2PO4+12.5 mmol/L庚烷磺酸鈉(pH5.0)/甲醇(50∶50,V/V);流動相總流速:0.06 mL/min;分離電壓:1 kV;檢測波長220 nm;分流閥壓力調至10.5 MPa。
1.3 數據處理
采用TriSepTM-2100加壓毛細管電色譜系統(上海通微公司)采集數據,Microsoft Excel 2010和Origin 8.0軟件進行數據處理與分析。
2 結果與分析
2.1 pCEC色譜條件的優化
2.1.1 檢測波長的選擇 用紫外分光光度計在200~400 nm范圍內分別對濃度均為80 μg/mL的綠原酸、阿魏酸、表兒茶素標準溶液進行掃描,結果如圖1所示,三種多酚在220 nm處都有較強的紫外吸收,因此選擇220 nm作為檢測波長。
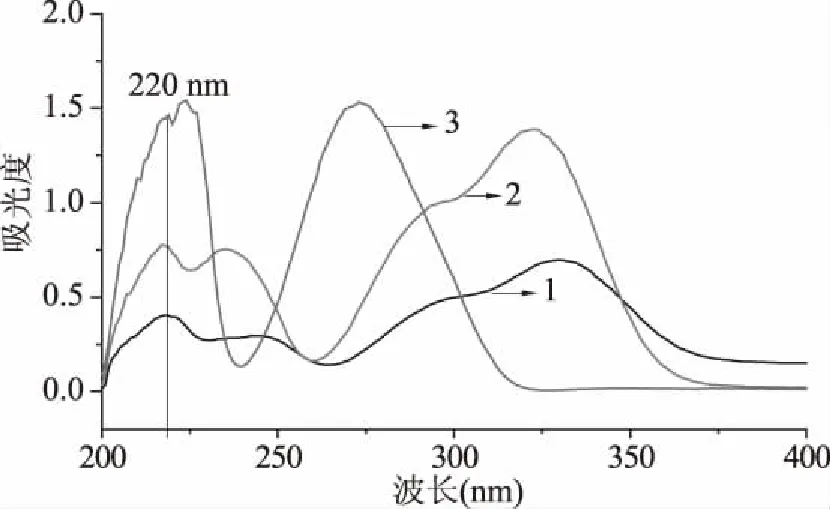
圖1 紫外吸收色譜圖Fig.1 Ultraviolet absorption chromatogram注:1:綠原酸;2:阿魏酸;3:表兒茶素;圖2~圖9同。
2.1.2 有機相比例的優化 有機相在流動相中所占比例會影響樣品在固定相和流動相之間的分配比例,進一步改變分離選擇性。實驗考察了甲醇比例分別為60%、55%、50%、45%及40%對綠原酸、阿魏酸、表兒茶素分離的影響。取“1.2.2”所配混合標準溶液,按照“1.2.4”所述 pCEC色譜條件進行分離檢測(圖2~圖8均在此條件下檢測)。如圖2所示,隨著甲醇比例為逐步降低,三種多酚類化合物的出峰時間逐步增加,60%甲醇時,綠原酸和阿魏酸分離度較低且表兒茶素峰型較差;55%甲醇時,表兒茶素峰形拖尾;50%甲醇時,三種多酚可得到較好的基線分離,保留時間較短,峰形較好,理論塔板數較高;隨著甲醇比例的進一步降低,即甲醇比例為45%和40%時,三種多酚類化合物保留時間較長,不利于快速分離檢測。因此,實驗選擇50%甲醇作為最優有機相比例。
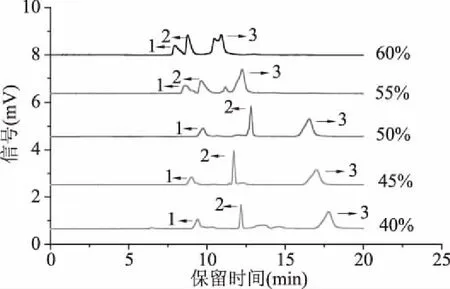
圖2 不同甲醇比例電色譜圖Fig.2 pCEC electropherograms at different methanol ratios
2.1.3 緩沖溶液種類的優化 pCEC分離系統中,為了形成穩定的電場和電滲流,需要在流動相中加入可以導電的酸堿或者緩沖鹽溶液。實驗分別考察了濃度均為15 mmol/L的CH3COONa、CH3COONH4、NaH2PO4緩沖溶液對上述三種多酚類化合物的分離影響。如圖3所示,當流動相中加入CH3COONa緩沖溶液時,表兒茶素峰形前沿,峰寬較寬;流動相中加入CH3COONH4緩沖溶液時,綠原酸、阿魏酸、表兒茶素均峰形拖尾或前沿,柱效較低;而采用NaH2PO4緩沖溶液時,三種多酚類化合物的峰形得到明顯改善,減輕或消除了拖尾或前沿現象,峰形較為對稱,柱效較高。因此,實驗選擇NaH2PO4作為流動相中添加的緩沖鹽。
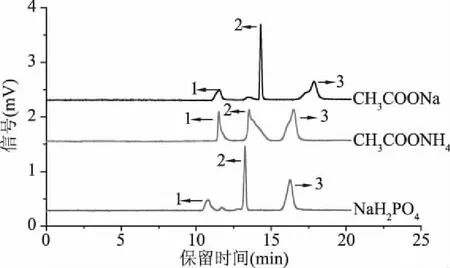
圖3 不同緩沖鹽電色譜圖Fig.3 pCEC electropherograms at different buffer salt
2.1.4 緩沖溶液濃度的優化 緩沖鹽溶液濃度決定了Zeta電位、雙電層厚度和毛細管內壁表面電荷超量等,對樣品的分離、樣品峰柱效等有著明顯的影響,因此,實驗進一步考察了NaH2PO4緩沖溶液濃度分別為10.0、12.5、15.0、17.5、20.0 mmol/L時對綠原酸、阿魏酸、表兒茶素分離的影響。結果如圖4所示。隨著NaH2PO4緩沖溶液濃度的增加,三種多酚峰形改善、柱效升高,但當NaH2PO4濃度過高時,電流值增大,產生較高的焦耳熱,反而造成峰形拖尾,柱效下降。綜合考慮,實驗選擇15.0 mmol/L NaH2PO4作為最優緩沖鹽濃度。
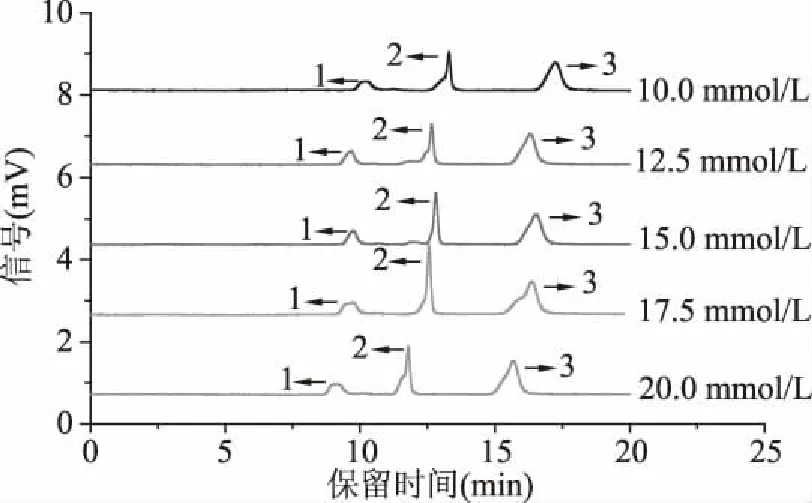
圖4 不同濃度NaH2PO4電色譜圖Fig.4 pCEC electropherograms of NaH2PO4 at different concentrations
2.1.5 離子對試劑濃度的優化 由于綠原酸、阿魏酸、表兒茶素以及甘蔗中干擾成分結構相似,含有較多酚羥基,且甘蔗中干擾成分眾多,均導致了待測樣品分離難度。而離子對試劑可通過與帶電荷分析物結合,形成配對離子,從而選擇性增加帶電荷分析物的保留時間,因此在緩沖鹽溶液中加入一定濃度的離子對試劑,可改善三種多酚之間以及多酚與甘蔗中其它干擾成分的分離。實驗采用庚烷磺酸鈉作為離子對試劑,并考察了當庚烷磺酸鈉濃度分別為5.0、7.5、10.0、12.5、15.0 mmol/L時的三種多酚的分離情況。如圖5所示,隨著庚烷磺酸鈉濃度的升高,三種多酚的分離度越來越好,保留時間逐步縮短,當庚烷磺酸鈉濃度為15.0 mmol/L時,阿魏酸、表兒茶素峰出現拖尾和前沿,分離度下降。綜合分離度和保留時間,選擇12.5 mmol/L庚烷磺酸鈉作為最優離子對試劑濃度。

圖5 不同濃度離子電色譜圖Fig.5 pCEC electropherograms of ion pair reagent at different concentration
2.1.6 流動相pH的優化 綠原酸、阿魏酸、表兒茶素都含有酚羥基,容易在溶液中電離,呈現出一定酸性。pH可通過改變三種多酚類化合物的電離情況,從而通過改變其電色譜行為,改變其分離情況。實驗考察了不同pH的NaH2PO4緩沖溶液對三種多酚的分離情況。如圖6所示,緩沖鹽溶液pH分別為3.5、4.0、4.5時,隨著pH的增大,三種多酚的保留時間逐步減小,三種多酚的分析周期仍較長,不利于其快速分離測定;pH5.0時,三種多酚保留時間進一步減小,但峰形和分離度很好;pH5.5時,由于產生過大的電滲流,分析周期最短,但表兒茶素峰型也受到影響。因此,選擇pH5.0作為流動相的最優pH。

圖6 不同pH下電色譜圖Fig.6 pCEC electropherograms at different pH
2.1.7 分離電壓的優化 隨著施加電壓的增大,會產生大量的焦耳熱,從而影響電滲流的大小,進一步影響柱效。實驗考察了分別在-3、-1、0、1、3 kV電壓下三種多酚的分離效果,結果如圖7所示。當施加負壓時,三種多酚保留時間較短,當施加電壓為-3 kV時,綠原酸與阿魏酸峰重疊。當施加電壓為-1 kV時,阿魏酸與表兒茶素峰無法完全分離;當施加正壓時,三種多酚保留時間延長,但分離度較負壓好。當施加電壓為1 kV時,三種多酚的分離度高且峰型較好;當施加電壓為3 kV時,阿魏酸峰型較差,柱效下降,且保留時間過長。因此,選擇1 kV作為最優分離電壓。

表1 綠原酸、阿魏酸、表兒茶素的線性范圍、回歸方程、相關系數(r)、檢出限Table 1 Linear range,regression equation,correlation coefficient(r)and LODs of chlorogenic acid,ferulic acid and epicatechin
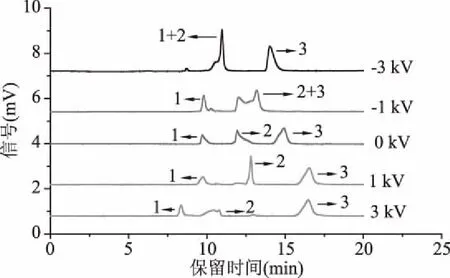
圖7 不同分離電壓下電色譜圖Fig.7 pCEC electropherograms at different voltages
2.1.8 流動相流速的優化 流速的大小直接影響了柱壓,從而影響各組分保留時間的長短以及分離情況,實驗考察了流動相流速分別為0.05、0.06、0.07 mL/min時綠原酸、阿魏酸、表兒茶素的分離情況。如圖8所示,隨著流動相流速的增大,三種多酚的保留時間縮短,分析時間減少,分離度下降。當流動相處于較低流速時,三組分峰形展寬,柱效下降;當流動相處于較高流速時,柱壓上升,表兒茶素峰型變差;在流速為0.06 mL/min時,三組分之間分離度好且仍能達到基線分離。因此,實驗選擇0.06 mL/min為最佳流動相流速。
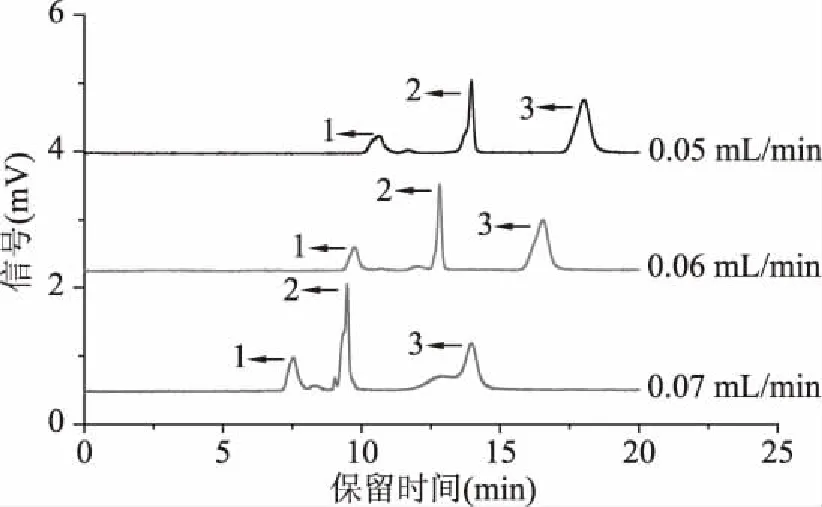
圖8 不同流速下電色譜圖Fig.8 pCEC electropherograms at different flow rates
2.2 方法學驗證
2.2.1 線性范圍和檢出限 取“1.2.1”所配制的標準溶液,按照“1.2.4”所述色譜條件進行分析,以質量濃度為橫坐標,峰面積為縱坐標繪制標準曲線。如表1所示,綠原酸、阿魏酸、表兒茶素在20~320 μg/mL范圍內線性關系良好,相關系數(r)在0.9922~0.9978之間,檢出限(LODs,S/N=3)分別為0.14、0.75、1.65 μg/mL。
2.2.2 精密度 分別取80 μg/mL的綠原酸、阿魏酸、表兒茶素標準溶液,連續進樣五次,結果顯示,綠原酸、阿魏酸、表兒茶素的相對標準偏差值分別為2.82%、2.69%、1.18%,表明儀器精密度良好。
2.2.3 樣品分析 將提取的樣品溶液,按照“1.2.4”所述 pCEC色譜條件進行分離檢測,結果如圖9。計算測得甘蔗渣中綠原酸、阿魏酸、表兒茶素的含量分別為135.68、99.66、21.98 μg/mL。

圖9 三種多酚混合標準溶液及甘蔗樣品電色譜圖Fig.9 pCEC electropherograms of three polyphenol mixed standard solutions and sugarcane samples注:a:混合標準溶液;b:樣品。
2.2.4 回收率 對樣品中的綠原酸、阿魏酸、表兒茶素進行加標回收率測定,結果如表2所示,綠原酸、阿魏酸、表兒茶素的回收率均良好,在96.79%~100.71%之間,RSD為2.24%~3.09%,表明該方法具有較好的準確度和重現性,適合甘蔗中多酚類化合物的分離檢測。
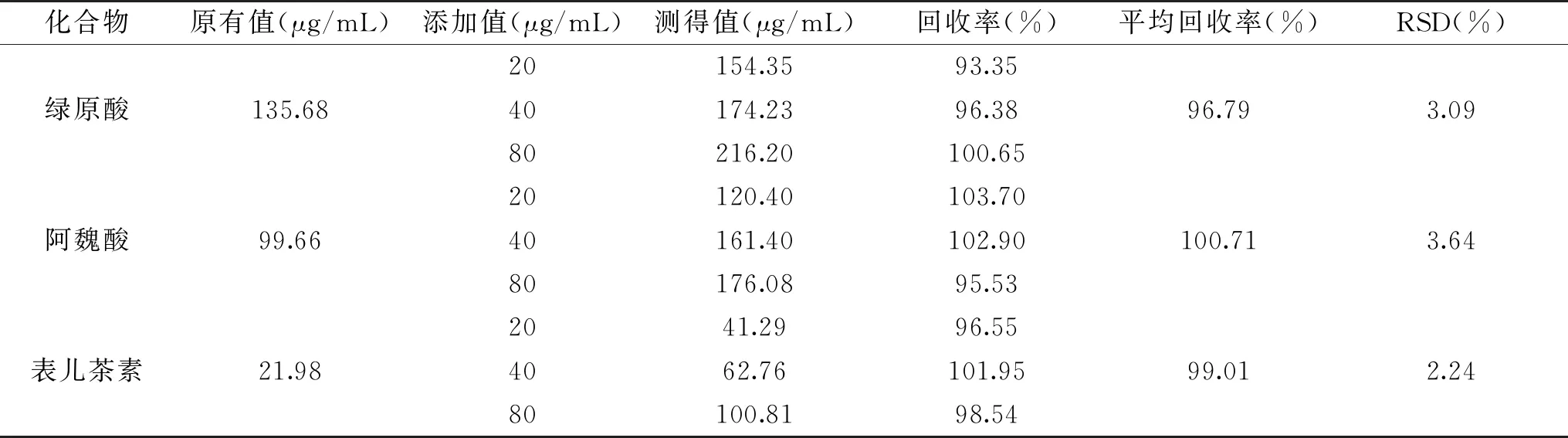
表2 甘蔗中綠原酸、阿魏酸、表兒茶素加標回收率(n=3)Table 2 Recovery of chlorogenic acid,ferulic acid and epicatechin in sugarcane(n=3)
3 結論
本實驗建立了加壓毛細管電色譜分離甘蔗中綠原酸、阿魏酸、表兒茶素的方法,優化了流動相比例、pH、緩沖鹽濃度、分離電壓、流速等色譜條件。在流動相為15.0 mmol/L NaH2PO4+12.5 mmol/L庚烷磺酸鈉(pH5.0)/甲醇(50∶50,V/V);分離電壓為1 kV;流動相總流速為0.06 mL/min;檢測波長為220 nm的色譜條件下,綠原酸、阿魏酸、表兒茶素在線性范圍為20~320 μg/mL的檢出限分別為0.14、0.75、1.65 μg/mL,加標回收率為96.79%~100.71%,RSD為2.24%~3.09%。該方法快速、高效,可用于甘蔗中綠原酸、阿魏酸、表兒茶素的分離檢測。
猜你喜歡
中國設備工程(2022年12期)2022-07-11 04:33:00
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2020年12期)2021-01-18 06:57:46
中學生數理化·七年級數學人教版(2020年12期)2021-01-18 06:57:46
中學生數理化·七年級數學人教版(2019年9期)2019-11-25 07:34:36
中學生數理化·七年級數學人教版(2019年9期)2019-11-25 07:34:34
中學生數理化·七年級數學人教版(2019年12期)2019-05-21 02:53:50
中學生數理化·七年級數學人教版(2019年12期)2019-05-21 02:53:48
