氟丙嘧草酯原藥高效液相色譜分析
2020-03-19 06:57:02張繼偉牛洺鑫王鵬思
世界農藥 2020年2期
張繼偉,牛洺鑫,王鵬思,張 博
(中化化工科學技術研究總院有限公司,北京 100083)
氟丙嘧草酯(butafenacil,CAS登錄號134605-64-4)是先正達公司開發的非選擇性脲嘧啶類除草劑,商品名稱Inspire,其混劑Rebin GT為氟丙嘧草酯+草甘膦。試驗表明,氟丙嘧草酯作為非選擇性除草劑,對于某些雜草有優異的防效[1]。雖然目前氟丙嘧草酯原藥和相關制劑還沒有在國內登記,但是對原藥的工藝研究和生物活性研究一直在進行[2-7],由此可見,該產品在國內的關注度較高,未來有望得以登記并商業化。本文采用高效液相色譜法對氟丙嘧草酯原藥的定性、定量分析進行了研究,首次公開報道了一種快速、簡便、準確的氟丙嘧草酯原藥分析方法,為后期該產品及其相關制劑配方的分析研究提供理論依據。
1 試驗部分
1.1 材料與試劑
99.0 %氟丙嘧草酯標樣(德國 Dr.Ehrenstorfer公司);水:純凈水(屈臣氏);乙腈:色譜純(Thermo Fisher);醋酸:分析純(國藥集團);氟丙嘧草酯原藥樣品(北京穎泰嘉和生物科技有限公司)。
1.2 儀器與設備
高效液相色譜儀:安捷倫1200高效液相色譜儀,配有紫外檢測器和自動進樣器;色譜柱:Zorbax Eclipse XDB-C18色譜柱(150×4.6 mm,5μm);超聲波清洗器(昆山市超聲儀器有限公司);0.45 μm有機相針式過濾器(津騰);50 mL容量瓶(北京化玻站有限公司)。
1.3 液相色譜操作條件
流動相:乙腈-0.5%醋酸水溶液(體積比60.0∶40.0);流速:1.0 mL/min;檢測波長:272 nm;柱溫:30 ℃;進樣體積:5.0 μL。保留時間:約6.2 min。氟丙嘧草酯原藥的高效液相色譜圖見圖1。
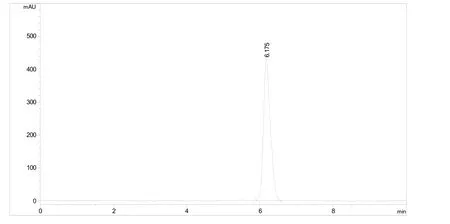
圖1 氟丙嘧草酯原藥的高效液相色譜圖
1.4 測定步驟
1.4.1 標樣溶液的配制
稱取0.05 g氟丙嘧草酯標樣(精確至0.000 2 g),置于50 mL容量瓶中,加入適量乙腈,在超聲波清洗器上超聲10 min,使標樣全部溶解,冷卻至室溫后,用乙腈準確定容至標線,搖勻,用0.45 μm濾膜過濾,備用。按照上述方法分別配制標樣溶液1和標樣溶液2。
1.4.2 試樣溶液的配制
稱取0.05 g(精確至0.000 2 g)氟丙嘧草酯原藥試樣,置于50 mL容量瓶中,加入適量乙腈,在超聲波清洗器上超聲10 min,使試樣全部溶解,冷卻至室溫后,用乙腈準確定容至標線,搖勻,用0.45 μm濾膜過濾,備用。按照上述方法分別配制試樣溶液1和試樣溶液2。
1.5 測定
在上述操作條件下,待儀器基線穩定后,連續進數針標樣溶液,直至相鄰2針氟丙嘧草酯標樣的響應值相對變化小于1.5%后,按照標樣溶液1、試樣溶液1、試樣溶液1、標樣溶液1、標樣溶液2、試樣溶液2、試樣溶液2、標樣溶液2的順序進行測定。
1.6 計算
將測得的2針試樣溶液中氟丙嘧草酯峰面積以及試樣前后2針標樣溶液中氟丙嘧草酯響應因子分別進行平均。試樣中氟丙嘧草酯的質量分數ω1(%)按式(1)和(2)計算:

式中:f為標樣溶液中氟丙嘧草酯的響應因子;m1為氟丙嘧草酯標樣的質量,g;A1為標樣溶液中氟丙嘧草酯峰面積的平均值;ω1為試樣中氟丙嘧草酯的質量分數,%;A2為試樣溶液中氟丙嘧草酯峰面積的平均值;ω為氟丙嘧草酯標樣的純度,%;m2為試樣的質量,g。
將試樣溶液1和試樣溶液2中測得的氟丙嘧草酯質量分數的平均值作為測定結果。
2 結果與討論
2.1 檢測波長的選擇
在190~380 nm的波長范圍內,對氟丙嘧草酯進行紫外掃描,得到其相應的吸收波長與響應值的紫外吸收光譜圖。發現氟丙嘧草酯在 272 nm波長有最大紫外吸收,雜質干擾小,且流動相無吸收。因此,綜合考慮多種因素后,將檢測波長選定為272 nm。
2.2 流動相的選擇
采用安捷倫Zorbax Eclipse XDB-C18(150×4.6 mm,5μm)色譜柱進行反相液相色譜分析。依據氟丙嘧草酯自身的理化性質,采用溶解性能較好的乙腈-水體系作為流動相。試驗結果表明當僅使用乙腈-水體系時,檢測峰形有拖尾現象,故在水相中加入了千分之五的醋酸來調節流動相pH,以便改善出峰拖尾現象。通過對不同配比的乙腈-0.5%的醋酸水體系的分離效果對比,確定最佳的流動相配比為乙腈-0.5%醋酸水體積比60.0∶40.0,流速為1.0 mL/min。此時,有效成分的保留時間為6.2 min左右,檢測峰形尖銳且對稱,沒有拖尾現象。此方法達到了快速簡便分離的要求,是一種理想的氟丙嘧草酯原藥分析方法。
2.3 方法線性相關性的測定
以乙腈為溶劑,分別配制質量濃度約為0.400g/L、0.700 g/L、1.000 g/L、1.100 g/L、1.300 g/L、1.600 g/L的氟丙嘧草酯標樣溶液。按照上述色譜條件,依次進行測定。將測得的結果,以進樣質量濃度為橫坐標,不同質量濃度溶液的峰面積為縱坐標,繪制氟丙嘧草酯的線性相關曲線。結果表明:氟丙嘧草酯在0.400~1.600 g/L呈良好的線性關系,其線性方程為y=5 585.1x+35.178,線性相關系數R2為0.999 8。
2.4 方法精密度的測定
在上述色譜分析條件下,從同一個氟丙嘧草酯原藥樣品中準確稱取6個試樣,進行平行測定,考察方法的精密度,試驗結果見表1。氟丙嘧草酯的標準偏差為0.11,變異系數為0.11%。結果表明該方法的重現性良好,精密度高,能夠滿足定量分析的要求。
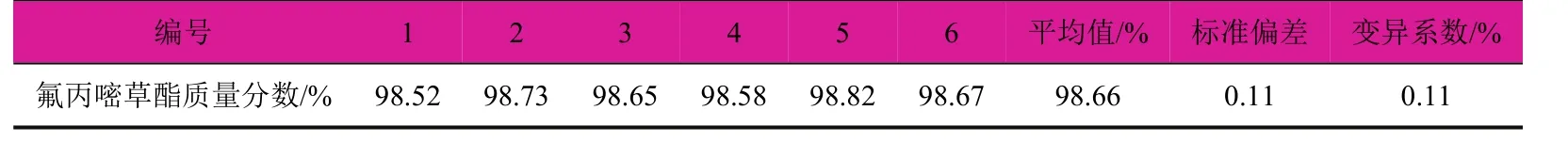
表1 精密度試驗
2.5 方法準確度的測定
準確稱取6個已知質量分數的氟丙嘧草酯原藥樣品,分別加入一定量的氟丙嘧草酯標準品,按照1.3章節的液相色譜條件測定待測樣品中氟丙嘧草酯的質量,計算其回收率,結果見表2。氟丙嘧草酯的平均回收率為99.94%。試驗結果表明該方法回收率較好,準確度高。
3 結 論
采用反相高效液相色譜法對氟丙嘧草酯原藥進行了定性定量分析。試驗結果表明:該方法簡單快速,分離效果好,峰形尖銳對稱,線性關系好,精密度和準確度高,適用于氟丙嘧草酯原藥的定量分析,也為該產品相關制劑配方的分析研究提供了參考。
猜你喜歡
中學生數理化·八年級物理人教版(2021年12期)2021-12-31 03:23:08
中學生數理化·中考版(2020年10期)2020-11-27 01:59:48
中國生殖健康(2019年2期)2019-08-23 08:12:08
兒童故事畫報(2019年5期)2019-05-26 14:26:14
產品可靠性報告(2017年7期)2017-09-05 09:49:12
Coco薇(2016年2期)2016-03-22 02:42:52
汽車觀察(2016年3期)2016-02-28 13:16:26
Coco薇(2015年1期)2015-08-13 02:47:34
小雪花·成長指南(2015年7期)2015-08-11 15:03:12
小雪花·成長指南(2015年4期)2015-05-19 14:47:56
