歐盟農藥原藥審批和制劑登記簡介
2020-03-19 06:56:58申繼忠
世界農藥 2020年2期
申繼忠
(1.上海艾農國際貿易有限公司,上海 220122;2.江蘇耘農化工有限公司,江蘇鎮江212132)
1 原藥審批程序
歐盟農藥登記分兩個步驟,一是在歐盟層面的原藥審批,二是在成員國層面的制劑產品登記。
歐盟層面原藥審批程序可簡述如下:(1)首先向歐盟某成員國提交原藥審批申請,該成員國被稱為起草評審報告的成員國(RMS)或主審國;(2)RMS確認原藥審批之申請是否可以被接受,這需要45 d,如需要提交額外信息,另加3個月時間;(3)RMS準備評審報告草稿(DAR),通常需要12個月,如果需要額外信息,另加6個月;(4)RMS提交DAR給歐盟食品安全管理局(EFSA),再由EFSA將DAR分發給申請人和其他成員國征求評論意見,此過程一般需要2個月;(5)EFSA做出風險評價結論,一般需要4個月;如果需要咨詢專家,另加最多1個月;如果需要提交額外信息,則另加5個月;(6)歐盟委員會接受風險評價結論,并經植物、動物、食品和飼料常務委員(SCoPAAFF)投票決定是否批準原藥審批申請;最后在歐盟官方雜志上以法規(Regulation)的形式發表結論;此過程需時6個月。
根據歐盟農藥新法規(指Regulation(EU)No.1107/2009,以下同),對1個新農藥有效成分(原藥)的審批,從申請被接受之日起到結論在官方雜志發表需要2.5~3.5年。這個時間是不確定的,實際長度決定于評審的復雜性和資料(卷宗)的齊全程度。在2011年6月14日之前提交的并被認為是完整的申請卷宗則遵守稍微不同的另一個評審程序(即依據法規Regulation 188/2011)。
歐盟原藥審批所需資料主要包括:產品鑒定、理化性質和技術特性、分析方法、藥效、毒理和代謝、作物中的殘留、環境歸宿和生態毒理等。
申請原藥審批時,須同時提交1個代表性制劑產品的全套資料。
2 原藥審批的續展
2.1 原藥審批續展程序
歐盟農藥新法規第14-20款設定了有效成分在10年有效期過后的續展申請程序,續展程序與初次審批程序類似。RMS和EFSA分擔風險評價責任。歐盟發布了續展程序指導文件,以保證申請人進行的研究和測試能遵守統一標準,并保證RMS和EFSA層面風險評價程序的一致性。
歐盟委員會最后負責向SCoPAFF遞交評審報告(review report)和執行法規草案(implementation regulation draft)。由SCoPAFF投票決定是否續展有效成分的有效期,最多延長5年。同意續展的,歐盟委員會采納執行法規并在歐盟官方雜志發表。續展申請一般要求在原藥審批到期之前提前3年提出。
主要步驟如下:(1)申請人準備并提交續展申請,所需時間不固定,決定于申請人;(2)RMS檢查申請材料的可接受性,需時1個月;(3)RMS進行評審并提交續展評價報告(RAR),需時11個月;(4)EFSA接收到RAR并傳遞給申請人和其他成員國,并收集他們的評論,需時2個月;(5)EFSA進行專家會議咨詢并做出最后結論,至少需要1個月時間,若需要補充材料,增加1個月,RMS評審補充的材料另需2個月時間;(6)EFSA做出最后結論,需時6個月;(7)歐盟委員會決議,由SCoPAFF投票決定是否批準續展,如批準,在官方雜志發表執行法規,需時6個月。
根據歐盟新法規第43款規定,在有效成分續展得到批準之后,擬續展含有該有效成分之制劑登記的,申請者須在有效成分續展后3個月內提出制劑續展申請,有可能需要提交新數據。
2.2 原藥審批續展
原藥審批獲得批準之后有效期為10年。在歐盟農藥新法規實行之前,依據歐盟理事會指令Directive91/414/EEC啟動了2個原藥審批續展計劃(active ingredient renewal programme,AIR)。第 1個計劃依據法規Regulation EC 737/2007(AIR-1)進行,第2個計劃是依據法規Regulation EU 1141/2010(AIR-2)進行。
AIR-2根據法規Regulation EU 1141/2010實施,包括31種原藥化合物。另有指導文件(Renewal Guidance on implementation of Regulation EU 1141/2010)規定了附加的程序指導。
在歐盟農藥新法規中,原藥續展規定體現在14-20款,不包括在AIR-1和AIR-2中的原藥。依據歐盟農藥新法規在法規Regulation EU 844/2012中給出續展規定。
AIR-3是針對有效期在2013年1月1日到2018年12月31日期間的有效成分150種。
有效成分審批的續展需要提出續展申請才能得到處理。根據法規Regulation EU 844/2012第3(6)款,歐盟委員會發表每個有效成分的續展申請人的名稱和地址。申請人的申請必須在法規Regulation EU 844/2012第1(1)款規定的日期之前提交并包括第2款規定的所有材料。歐盟文件SANCO/10148/2014對提交的所有申請進行了綜述。
AIR-4是針對有效期在2019年1月1日到2019年12月31日到期的有效成分。詳細內容可參見歐盟委員會執行決議Commission Implementing Decision 2016/C 357/05。主審國和輔審國(Co-RMS)之名單發表在法規Regulation EU 2016/183中。根據有效成分的最初有效期及有效成分的特性將它們劃分為4組,屬于低風險的有效成分以及不能滿足審批標準的有效成分予以優先評審,還有一部分有效成分需要延長審批有效期。
申請人必須提前3年提交擬續展的有效成分的續展申請。只有在歐盟委員會收到續展申請之后才能進行評審并延長有效成分的有效期。如果沒有收到申請,原有效期保持不變。
同樣,歐盟委員會也將發表每個有效成分的續展申請人的名稱和地址。申請人的申請必須依法及時提交申請和所需材料。文件SANTE-2016-11734對提交的所有申請進行了綜述。
第5次續展計劃針對有效期在2022年1月1日到2024年12月31日到期的有效成分。指定的RMS及輔審國名單發表在法規Regulation EU2018/155中。那些被批準可作為替代產品的有效成分將被優先評審,并立即開始。其他一些有效成分有可能需要延長其有效期。工作計劃草案SANTE/2018/10048發表了這些有效成分的有效期、應該提交申請的日期、應該提交補充材料的日期等詳細內容。
續展步驟如下(與新有效成分審批相似):(1)準備和提交申請及補充卷宗(需時不固定,決定于申請人);(2)RMS審查卷宗可接受性(需時1個月);(3)RMS進行評價并提交評價報告(RAR),需時11個月;(4)EFSA接收到RAR并將RAR分發給各成員國及申請人征求評論,需時2個月;(5)EFSA召開專家咨詢會并做最后結論,需時至少1個月;若需要增加資料,另加1個月;評審增加資料另加2個月;(6)EFSA最后做出結論,需時6個月;(7)SCoPAFF就歐盟委員會建議投票,并發布最后的執行法規,需時6個月。
與新原藥審批類似,現有原藥的續展也需要約2.5~3年。
3 原藥等同性認定(中國原藥進入歐盟的重要渠道)
不同來源或不同方法生產的相同的有效成分(原藥),如果其對環境或健康的危害水平與現有的已獲得批準的原藥(參比源)相當,那么新來源或新方法生產的原藥可被認為與現有的已獲批準的原藥等同。如果新來源的原藥比參比源原藥毒害更大,那么必須對其進行風險評價以確定含有該新來源原藥的制劑產品能夠滿足歐盟農藥新法規規定的安全標準要求。
歐盟農藥新法規第38款說明了原藥等同性評價過程,針對化學農藥和微生物農藥分別有等同性認定指導文件可供參考。
對于普通原藥(已無專利保護或專有使用權保護),如果某有效成分(原藥)已經在歐盟獲得批準,新來源的含有相同有效成的原藥也必須獲得審批之后才能上市。
新來源的原藥首先要經過等同性認定,即由歐盟成員國將新來源的原藥之技術標準與現有來源的原藥或已經列入新法規之附錄1的原藥(參比源)之技術標準進行對比。
在進行等同性認定之前,成熟和穩定的生產工藝保證是非常必要的。等同性認定報告會給出工藝過程不穩定或工藝過程控制良好2種評審結果。
等同性認定分2個步驟進行:第1步是化學等同性認定,如果化學不等同則需要進行第2階段的毒理學等同性認定。
原藥等同性認定要求的資料包括:
(1)原藥5-批次GLP全分析報告。試驗所用原藥樣品必須是來自近期的大生產,而且要采自非連續的批次。目的是使樣品能代表生產過程的穩定性水平。
(2)申請人要從該5-批次全分析報告制定出產品技術標準。該標準要反應生產過程的變化性。標準內容包括有效成分含量和任何含量>0.1%(w/w)的雜質的最低含量。
(3)對所有含量>0.1%(w/w)的雜質都必須進行定性鑒定和定量分析。此外,尤其是那些由于毒性問題、生態毒性問題或環境問題而不受歡迎的雜質應該被視為相關雜質。相關雜質的限量可能設定在低于0.1%的水平,而且分析方法必須根據指導文件SANCO/3030/99 Rv4進行驗證。
(4)新來源原藥有效成分含量不得低于參比源的含量,有效成分含量和雜質限量是標準的關鍵。
(5)提交技術標準時,還要提交保密信息文件,內容包括生產原料、詳細制造過程、雜質情況等。
(6)原藥等同性認定的申請須向某一成員國提交,但是其他所有成員國都有權對評審報告提出他們自己的意見,評價報告必須獲得同意才能通過評審。
(7)評審的目的是確定新來源原藥與參比原藥相比是否有不可接受的危害增加。因此需要明確:①是否有新的雜質;② 相關雜質的增加水平;③含量>0.1%(w/w)的非相關雜質的增加水平。
(8)如果能保證新來源之原藥有效成分最低含量不比參比源低,而且沒有新雜質或現有雜質含量也沒有增加,則新來源原藥可被認為與參比源等同。
(9)如果出現新雜質或現有重要雜質或相關雜質含量增加超過可接受水平,那么可能需要提交更多毒理資料,資料要求視具體產品而定。所要求的資料可以是(Q)SAR分析報告、基因(遺傳)毒性試驗、科學論證和/或毒性數據。對于毒理學數據評價,專家的判斷很重要。
毒理學數據要求如表1所示。
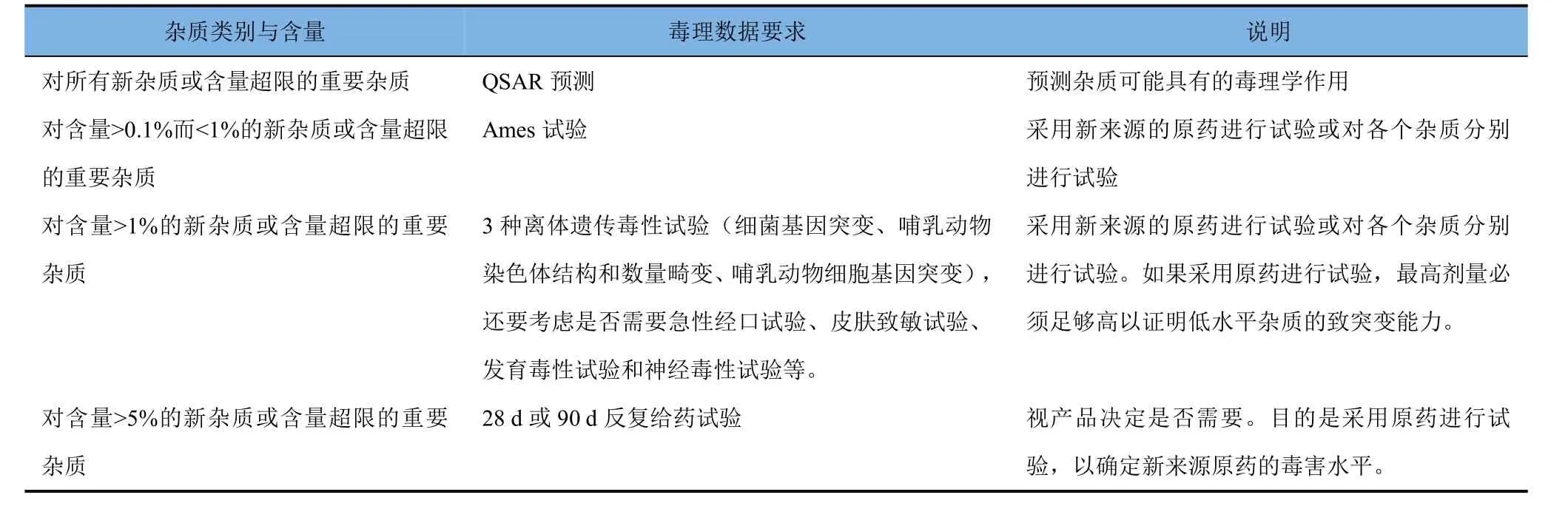
表1 毒理學等同性認定對毒理數據的要求
由于參比源產品的資料對新來源的申請人是保密的,因此申請人在提交申請之前無法知道等同性認定的結果。
在技術上被評估為等同是獲得歐盟批準的第1步。獲得等同性認定并不意味著可以將新來源的原藥立即投放到歐盟市場,除非能獲得必要的活性物質(原藥)數據。
因此,首先要能夠獲得對有效成分(原藥)數據(歐盟農藥新法規附錄II要求的數據)的訪問權。訪問授權的獲得有不同的方式,如通過付費或不付費獲得數據訪問授權書(Letter of Access,LOA),或參考過保護期的數據,或者有潛力的申請者自己做出相同的數據卷宗(雖然成本較高,但是有更長的銷售時期)。有潛力的申請者還要考慮進行中的有效成分續展計劃在未來可能提交的新的可受保護的數據。
在大多數歐盟成員國,新來源必須書面通知相關的政府主管部門。有些成員國要求將擬用于制劑產品的新來源原藥正式予以授權。
中國農藥原藥產品擬出口歐盟市場的,首先須經過等同性認定。
4 歐盟農藥制劑(PPP)登記
歐盟農藥制劑分區登記體系是為了保證產品在各成員國的登記能協調有效地進行。
歐盟分3個區,即南、中和北區。歐盟國家可以代表本區內的其他國家評價PPP登記申請,有時也能代表所有3個區。歐盟農藥新法規制定了PPP登記要求、程序和時間表。
申請者、歐盟成員國、歐盟委員會和歐洲食品安全局(EFSA)可以參與PPP登記評審過程。
根據PPP的預期用途、需要PPP的成員國以及任何現有產品登記的監管狀況,可以提交不同類型的申請。
申請人如欲在歐盟獲得PPP登記,必須通過植保產品申請管理系統(PPPAMS)向成員國遞交申請。這個在線系統管理申請的工作流程,將相關信息上載到一個歐盟范圍內登記PPP的公共數據庫。
數據卷宗不通過PPPAMS提交,在每個歐盟國家的其他手工流程(例如開發票/收費和評估工作)也不通過PPPAMS。
PPPAMS管理PPP登記申請的工作流程,使申請人和歐盟國家能夠在申請過程中彼此溝通,主要是通過更改申請所處的狀態達到彼此了解申請進程。
PPPAMS可以在申請過程中根據申請狀態的變化情況發通知、發送信息并采取行動等。
PPPAMS并沒有取代歐盟國家現有的申請程序,它與歐盟國家在PPPAMS之外運行的其他電子和手工流程聯合工作。在下面解釋PPP的不同申請類型的表格中,說明了哪些步驟在PPPAMS內部或外部實現。每個成員國都有自己的程序,可以從網絡上獲取。
以下分別介紹PPP各種類型的登記。
4.1 PPP首次登記
歐盟農藥新法規之28-39款解釋了PPP登記的要求、內容和程序。
PPP登記申請是按區域進行評估的,但對于某些用途的PPP,歐盟被認為是一個單一區域,某一個成員國便可以代表整個歐盟對該PPP進行評估。這些用途是指:溫室用、收獲后處理、空的儲藏室或容器的處理、種子處理等。因為這些用途不受環境、氣候和農業生產條件等的影響。
新的PPP登記程序和之后在歐盟各成員國之間的互認程序可簡述如下。
申請人須通過PPP申請管理系統(PPPAMS)提出申請將PPP投放到歐盟某個國家或某些國家之市場。每一個擬投放產品的區域都選擇1個該區域內的主審國(zRMS)(如前所述有些用途則由1個成員國代表所有區域進行評估)。
(1)zRMS對申請進行評估;
(2)區域內其他成員國對zRMS的評估發表評論;
(3)zRMS決定是否登記或拒絕登記;
(4)其他成員國決定給予或拒絕登記;
(5)如獲登記,而其后申請人希望將同一產品在其他成員國上市的,則須在有關成員國申請“互認”該產品的登記。
申請過程中的某些工作內容是由成員國以手工或電子程序在申請管理系統(PPPAMS)之外進行管理和處置。表2顯示了首次登記PPP的申請程序是如何工作的。
表1說明了首次申請PPP登記時,申請人是如何通過PPP申請管理系統向國家主管部門提交申請的。這個流程按照步驟很容易操作,它顯示了整個完整的申請流程,并突出顯示了在申請管理系統(PPPAMS)之外需要做什么,在PPPAMS之內需要完成什么,以及在流程的每個階段誰來做什么。
首次進行PPP登記申請,只可由申請人(行業/顧問公司用戶)提交。
在提交產品登記和向PPPAMS提交申請之前的任何階段都可以與zRMS/NCA(主管當局)進行提交前討論,包括會議和其他電子形式的討論。
如屬分區申請,申請人須填妥分區申請通知書表格(載于區域評價和相互承認指導文件之附錄3),并于提交前6個月確定zRMS。

表2 PPP登記首次申請流程
申請PPP登記需要提交的申請文件見表3。

表3 PPP申請需提交的文件(以英國為例)
4.2 區域間互認
互認使現有登記的持有人將同樣的產品以同樣的用途在不同區域同等農業條件下的其他國家申請登記。
歐盟農藥新法第40-42款解釋相互承認的規定、內容及程序,另有指導文件提供了有關該過程的詳細信息。
只有在現有的產品登記下,才可在其他成員國申請互認。對于通過申請管理系統獲得登記的產品,還可以通過該系統(PPPAMS)申請互認登記。
申請程序的某些部分由成員國經手工或電子程序在PPPAMS之外進行管理和處理。表4顯示了應用程序如何工作,以實現PPP的互認。
表4顯示了如何向PPPAMS國家主管部門遞交互認申請,這個流程按照步驟很容易進行。表4顯示了整個申請步驟和說明,并突出了需要在PPPAMS外部完成的工作以及需要在PPPAMS中完成的內容以及誰在這個過程的每個階段做什么。
在產品登記申請提交之前的任何階段,申請人與區域報告成員國/國家主管當局(NCA)之間都可以進行互認登記申請提交之前的初步討論,包括會議和其他電子形式的討論。

表4 區域互認流程表
4.3 修訂或撤銷現有的PPP登記
修訂PPP登記須遵循與前述首次登記相同的規定及程序。申請人可選擇隨時撤回登記,撤回請求應提交給有關成員國。如果有跡象表明不再滿足登記要求,成員國可以隨時審查登記。修改或撤銷現有的登記將在未來通過PPPAMS完成。
4.4 PPP登記續展
原藥被批準續展后,所有含有該原藥的植保產品也必須進行續展評估,以確保產品符合對原藥的最新評估,并具有新的科學和技術依據。PPP續展在區域層面上執行,歐盟農藥新法規第43款概述了申請的程序和時間表,而隨附的指導文件則為這類申請提供進一步的詳細資料。
修改或撤銷現有的登記也將通過PPPAMS完成。
4.5 PPP應急登記
歐盟農藥新法規第53款規定,在特殊情形下成員國可以最多120 d的限制使用和管制使用登記某些產品。這種特殊情形是指遇到任何其他措施無法控制的某種嚴重危險時需要的應急登記。
在批準應急登記時,需要通知歐盟委員會和所有其他成員國,歐盟委員會可以向歐洲食品安全管理局(European Food Safety Authority)征求意見或尋求科學/技術援助。
應急登記申請應通過PPPAMS遞交,這些申請要么由業界提出,要么由成員國提出(如果申請人是種植者、貿易組織或成員國地方當局)。有指導文件提供關于必須提供哪些信息來支持應用程序的進一步細節。
申請程序的某些部分由成員國經手工或電子程序在PPPAMS之外進行管理和處理,表5列出了申請緊急登記的過程。
表5說明了申請應急登記的人是如何通過PPPAMS向NCA提交申請的,申請人或使用者通常有行業或顧問資料,但有時NCA可以代表由于某種原因沒有自己帳戶的個人提交應急登記申請。必須指出,NCA只能提交用于應急使用的產品。對于所有其他類型的申請,產品只能由申請人(業界/咨詢用戶)自己創建。
開始處理應急申請需要注意的重要事項:對于應急登記申請,NCA可以繞過其他類型申請所需的正常狀態,使NCA能夠更快地處理應急申請。在PPPAMS中登記產品和申請之前,申請人和NCA可以進行初步的提交前討論,這些可以包括會議和其他電子形式的討論。

表5 PPP應急登記流程表

續表
4.6 小宗作物使用PPP申請
歐盟農藥新法規第51款,允許申請將小宗作物應用包括在現有的產品登記中,這項申請將來可以通過PPPAMS提交。
4.7 PPP平行貿易許可證
歐盟的主要支柱之一是在歐盟內部市場進行的貨物自由流通。
必須在不構成任何不必要障礙的情況下促進PPP在歐盟內部的自由流通,同時確保現行規定不會對人類健康、動物或環境構成任何風險。
平行貿易許可證允許在一個成員國(原登記成員國)登記的產品被引入另一個成員國(進口成員國),前提是該進口成員國確定有完全相同的產品已在其境內獲得登記。現有的簡化程序允許平行貿易登記在45 d內完成。
歐盟農藥新法規第52款解釋了平行貿易申請程序,成員國批準平行貿易許可證的3個基礎條件是:(1)提交申請的植保產品在原登記成員國已獲登記,而且該產品的參考產品在引進國也已獲得登記;(2)原登記成員國登記的產品與進口國的參考產品有相同的組成;(3)平行貿易申請須在進口國提交。
有平行貿易申請指南文件可供參考,未來可以通過PPPAMS申請平行貿易。
根據歐盟規則,從申請之日起至批準、修訂或撤回制劑產品登記書,最長需1.5年。這取決于申請的復雜程度和資料的完整程度以及申請的類型。
關于以上各項活動所需時間,這里給出英國的例子供讀者參考(表6)。
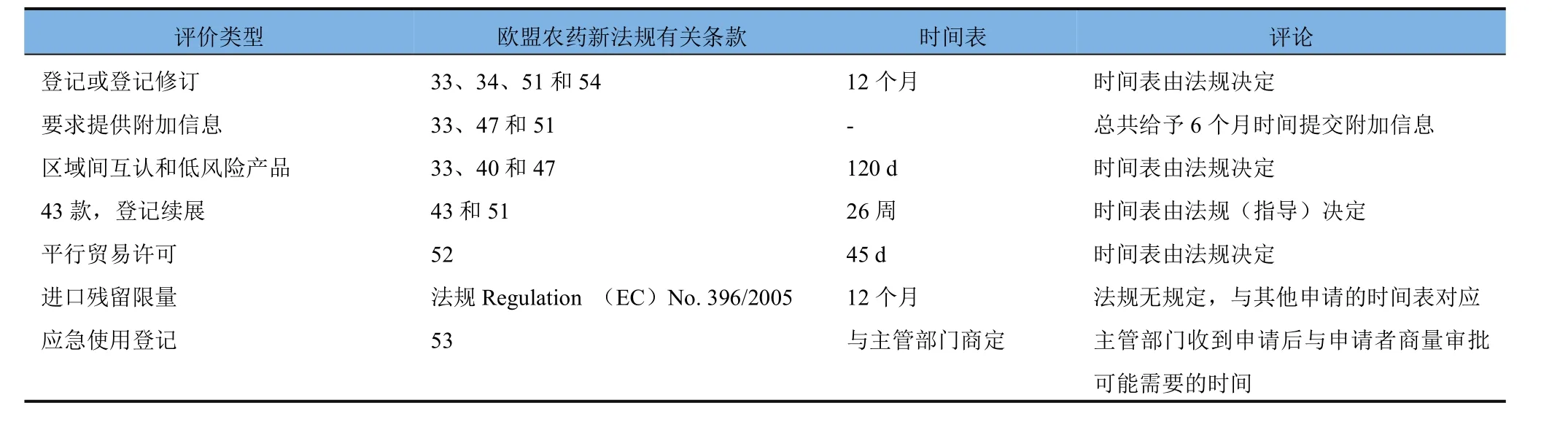
表6 英國新的PPP登記申請程序(始于2016年6月29日)下各種評價類型所需時間
總之,歐盟農藥之原藥審批或制劑登記都是一個漫長而細致的工作。本文只是簡單介紹,與實際操作可能有相當距離。國內企業一般都需要借助歐盟農藥登記專業咨詢公司或有能力的客戶去完成。希望本文能給讀者提供一定的背景知識,從而有利于讀者與咨詢公司溝通接洽。
猜你喜歡
現代裝飾(2022年4期)2022-08-31 01:39:32
現代裝飾(2022年3期)2022-07-05 05:55:06
人大建設(2019年12期)2019-05-21 02:55:44
中山大學法律評論(2018年1期)2018-03-30 01:21:00
瞭望東方周刊(2017年42期)2017-12-05 18:49:38
環球時報(2017-03-30)2017-03-30 06:44:45
中國衛生(2015年3期)2015-11-19 02:53:32
Coco薇(2015年1期)2015-08-13 02:23:50
政治與法律(2014年11期)2014-03-01 02:20:40
玩具(2009年10期)2009-11-04 02:33:14
