新型噻吩查爾酮衍生物的合成及其初步抗炎活性研究
2020-02-24 01:32:56唐燕玲楊小碧唐新羽杜文絨高金春毛澤偉
合成化學 2020年1期
唐燕玲, 張 霞, 楊小碧, 唐新羽, 杜文絨, 高金春, 毛澤偉
(云南中醫藥大學 中藥學院,云南 昆明 650500)
查爾酮是重要的黃酮類化合物,其化學結構為1,3-二苯基丙烯酮,廣泛分布在甘草、紅花等多種藥用植物中[1-3]。查爾酮含有獨特的α,β-不飽和酮單元,活性位點多,可與多種受體結合,表現出廣泛的生物活性。目前,已有多種查爾酮類化合物作為藥物用于臨床研究,如甲氧查爾酮作為利膽藥、索法酮作為抗潰瘍藥、橙皮苷甲基查爾酮和橙皮苷三甲基查爾酮可用于治療靜脈舒張等。查爾酮類化合物在新藥研發方面表現出的巨大潛力,使其成為藥物化學領域的研究熱點[4-6]。
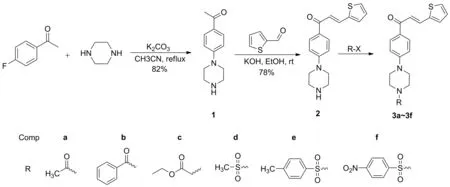
Scheme 1
查爾酮分子結構中含有A、 B兩個苯環,根據生物電子等排法的藥物設計原理,用芳雜環替換苯環,可能得到具有更優生理活性的化合物[7-10]。如Zheng等[11]發現噻吩、呋喃和喹啉查爾酮衍生物對變異鏈球菌具有較好的抑菌活性。Zhang等[12]發現含陽離子的噻吩、呋喃和吡啶查爾酮類衍生物對金黃色葡萄球菌、大腸桿菌和耐甲氧西林金黃色葡萄球菌等均有較強的抑制活性。在前期研究中,本課題小組發現哌嗪取代的呋喃查爾酮衍生物具有良好的抗炎和抗腫瘤活性[13-14]。基于此,如用噻吩替換呋喃環,有望得到生物活性更優越的噻吩查爾酮化合物。
本文以4-氟苯乙酮為原料,經取代、羥醛縮合及衍生化,合成了6個新型的噻吩查爾酮衍生物(3a~3f, Scheme 1),其結構經1H NMR,13C NMR和HR-MS(ESI)表征。以地塞米松作陽性對照,采用細菌脂多糖誘導小鼠巨噬細胞Raw 264.7炎癥模型對3a~3f的體外抗炎活性進行了測試。
1 實驗部分
1.1 儀器與試劑
Bruker AM 400 MHz型核磁共振儀(CDCl3為溶劑,TMS為內標);AutoSpec Premier P776 型雙聚焦三扇型磁質譜儀;Thermo-fishe二氧化碳培養箱;Epoch連續波長酶標儀。
所用試劑均為化學純;乙腈和二氯甲烷使用前經干燥處理。
1.2 合成
(1) 化合物1的合成
將4-氟苯乙酮1.38 g(10 mmol)、無水哌嗪1.72 g(20 mmol)和無水碳酸鉀2.76 g(20 mmol)加入50 mL圓底燒瓶中,加入干燥乙腈30 mL,回流反應過夜(TLC檢測)。攪拌下倒入200 mL冰水中,抽濾,濾餅用水(3×30 mL)洗滌,真空干燥得1的粗產物。不經純化,直接投入下一步反應。
(2) 化合物2的合成
將1的粗產物2.04 g(10 mmol)和2-噻吩甲醛1.12 g(10 mmol)加入100 mL圓底燒瓶中,加入甲醇30 mL和氫氧化鉀3.00 g,冰水浴冷卻,反應5 h(TLC檢測)。加入冷水60 mL,用二氯甲烷(3×20 mL)萃取,合并有機相,用無水硫酸鎂干燥,真空濃縮,剩余物用50%乙醇溶液重結晶得2的粗產品。無需純化,直接投入下一步反應。
(3)3a~3f的合成通法
稱化合物2的粗產物90 mg(0.3 mmol)加入15 mL圓底燒瓶中,依次加入無水DCM 10 mL,無水碳酸鉀138 mg(1 mmol)和鹵代物0.5 mmol,反應至終點(TLC檢測)。加入5%氫氧化鈉溶液10 mL,攪拌20 min,用二氯甲烷(3×10 mL)萃取,合并有機相,用無水硫酸鈉干燥,真空濃縮,殘余物經硅膠柱層析(洗脫劑:VMeOH/VDCM=2/98)純化得3a~3f。
化合物3a: 收率86%;1H NMRδ: 7.98(d,J=8.9 Hz, 2H), 7.92(d,J=15.3 Hz, 1H), 7.38(d,J=5.2 Hz, 1H), 7.35(d,J=15.2 Hz, 1H), 7.31(s, 1H), 7.05~7.07(dd,J=3.6 Hz, 3.7 Hz, 1H), 6.90(d,J=8.9 Hz, 2H), 3.78(t,J=5.0 Hz, 2H), 3.64(t,J=4.8 Hz, 2H), 3.40(t,J=5.4 Hz, 2H), 3.36(t,J=5.4 Hz, 2H), 2.13(s, 3H);13C NMR: 187.5, 169.2, 153.7, 140.8, 135.9, 131.6, 130.7, 129.0, 128.4, 128.3, 120.8, 114.0, 47.6, 47.3, 45.8, 41.0, 21.4; HR-MS(ESI)m/z: calcd for C19H20N2O2S[M+]340.1245, found 340.1240。
化合物3b: 收率80%;1H NMRδ: 7.99(d,J=8.8 Hz, 2H), 7.93(d,J=15.3 Hz, 1H), 7.30~7.42(m, 5H), 7.02~7.14(m, 4H), 6.91(d,J=8.9 Hz, 2H), 3.77(t,J=5.1 Hz, 2H), 3.63(t,J=4.9 Hz, 2H), 3.38(t,J=5.3 Hz, 2H), 3.35(t,J=5.4 Hz, 2H);13C NMR: 188.3, 169.7, 154.6, 147.9, 141.4, 140.2, 136.5, 135.1, 133.6, 132.1, 131.4, 130.0, 128.2, 127.5, 124.4, 120.4, 117.9, 115.3, 49.4, 47.0。
化合物3c: 收率87%;1H NMRδ: 7.98(d,J=9.0 Hz, 2H), 7.92(d,J=15.3 Hz, 1H), 7.31~7.38(m, 3H), 7.05~7.08(dd,J=3.6 Hz, 3.7 Hz, 1H), 6.91(d,J=9.0 Hz, 2H), 4.23(q,J=7.2 Hz, 2H), 3.44(t,J=5.0 Hz, 4H), 3.27(s, 2H), 2.75(t,J=5.1 Hz, 4H), 1.30(t,J=7.1 Hz, 3H);13C NMR: 187.6, 170.2, 154.1, 141.0, 135.7, 131.4, 130.7, 128.4, 128.3, 128.2, 121.1, 113.7, 60.9, 59.4, 52.7, 47.4, 14.4; HR-MS(ESI)m/z: calcd for C21H24N2O3S [M+]384.1508, found 384.1502。
化合物3d: 收率82%;1H NMRδ: 7.96(d,J=8.8 Hz, 2H), 7.92(d,J=15.3 Hz, 1H), 7.39(d,J=5.1 Hz, 1H), 7.34(d,J=15.3 Hz, 1H), 7.32(s, 1H), 7.00-7.06(dd,J=3.5 Hz, 3.6 Hz, 1H), 6.91(d,J=9.0 Hz, 2H), 3.43(t,J=4.9 Hz, 4H), 3.12(t,J=5.1 Hz, 4H), 3.01(s, 3H);13C NMR: 187.8, 153.4, 141.5, 133.8, 132.1, 130.9, 128.6, 128.0, 121.3, 113.7, 49.6, 47.1, 28.5。
化合物3e: 收率76%;1H NMRδ: 7.87(d,J=9.0 Hz, 2H), 7.85(d,J=15.3 Hz, 1H), 7.60(d,J=8.2 Hz, 2H), 7.22~7.31(m, 5H), 6.98~7.01(dd,J=3.6 Hz, 3.7 Hz, 1H), 6.79(d,J=9.0 Hz, 2H), 3.38(t,J=4.9 Hz, 4H), 3.09(t,J=5.2 Hz, 4H), 2.35(s, 3H);13C NMR: 187.6, 153.5, 144.2, 140.8, 136.1, 132.5, 131.6, 130.7, 129.9, 129.3, 128.4, 128.4, 128.0, 120.8, 114.1, 47.4, 45.8, 21.6; HR-MS(ESI)m/z: calcd for C24H24N2O3S2Na [M+Na+]475.1126, found 475.1122。
化合物3f: 收率69%;1H NMRδ: 8.06(d,J=8.8 Hz, 2H), 7.91(d,J=15.2 Hz, 1H), 7.54(d,J=5.3 Hz, 1H), 7.46(d,J=15.3 Hz, 1H), 7.39~7.46(m, 3H), 7.32(s, 1H), 7.09~7.13(m, 1H), 6.98(d,J=9.0 Hz, 2H), 3.39(t,J=5.4 Hz, 4H), 3.10(t,J=5.3 Hz, 2H);13C NMR: 188.0, 157.3, 145.6, 141.4, 139.4, 135.8, 133.2, 132.4, 130.0, 129.0, 128.7, 128.5, 124.5, 122.1, 114.2, 47.6, 47.5, 45.8, 41.2。
1.3 抗炎活性測試
以地塞米松(Dexamethasone)為陽性對照,采用細菌脂多糖(LPS)誘導小鼠巨噬細胞RAW264.7炎癥模型評價化合物抑制NO產生的活性。將對數生長期的RAW264.7細胞(1×106個/mL, 100 μL/孔)接種于96孔培養板中,用MTT法測定不同濃度化合物(40、 20、 10、 5、 2.5和1.25 μmol·L-1)的細胞毒性。確定無毒濃度后,取對數期生長的RAW264. 7細胞(2×106個/mL, 100 μL/孔)接種于96孔培養板中,設空白對照組,LPS模型組(加入終濃度為1.5 μg·mL-1的LPS),待測化合物組(同時加入藥物和終濃度為1.5 μg·mL-1的LPS),地塞米松組(終濃度為10 μmol·L-1地塞米松和終濃度為1.5 μg·mL-1LPS),于37 ℃, 5 %CO2中培養24 h,收集上清液,采用Griess法檢測炎性介質NO含量。每組實驗重復3次,并計算IC50。
2 結果與討論
2.1 合成
參考前期合成呋喃查爾酮的方法合成中間體2,發現收率較低。其原因可能是噻吩查爾酮活性較高,加熱時哌嗪會與α,β-不飽和酮發生邁克爾加成反應。隨后,我們對合成方法進行了調整,哌嗪和4-氟苯乙酮先經取代反應合成4-哌嗪苯乙酮(1),再與2-噻吩甲醛經羥醛縮合反應合成2,收率較高。
2.2 抗炎活性
表1為目標化合物的體外抗炎活性。由表1可以看出,化合物3d和3e能夠較好地抑制炎癥因子NO的生成,活性與地塞米松相當。從衍生物類型上來看,磺胺衍生物的抗炎活性強于酰胺和叔胺衍生物。在磺胺衍生物中,當苯環上含有供電子取代基時,活性較好(3e),當苯環上含有吸電子取代基時活性下降(3f)。
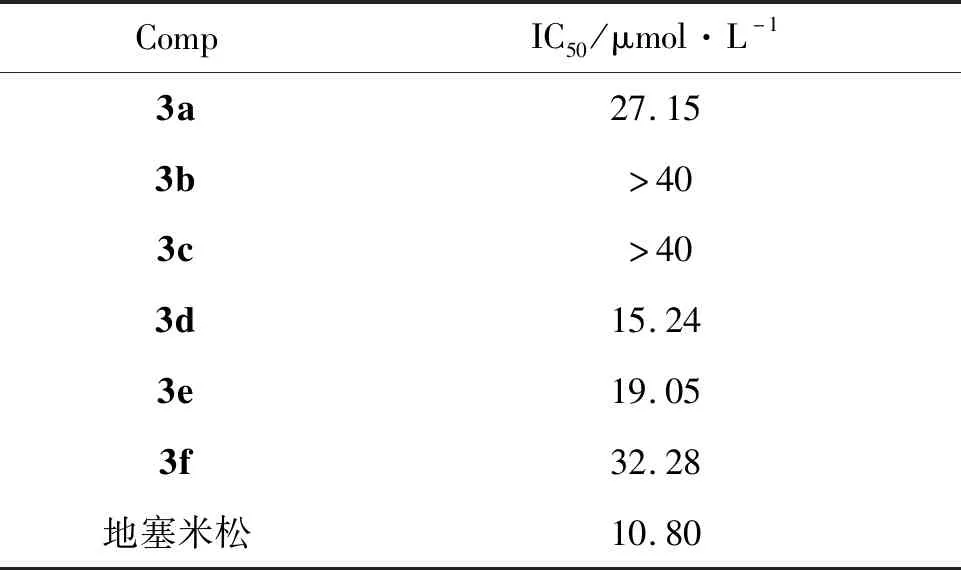
表1 3a~3f的體外抗炎活性
合成了6個新型的哌嗪取代噻吩查爾酮衍生物(3a~3f)。以地塞米松作陽性對照,采用細菌脂多糖誘導小鼠巨噬細胞Raw 264.7炎癥模型對3a~3f的體外抗炎活性進行了初步測試。結果表明:化合物3d和3e能有效抑制炎癥因子NO的生成(IC50分別為15.24 μM和19.05 μM)。
