Gilbert綜合征合并巨幼細胞貧血1例
2020-02-11 02:58:22康虹陽佟長青
中國現代醫學雜志 2020年1期
關鍵詞:基因突變
康虹陽,佟長青
(河北北方學院附屬第一醫院 血液科,河北 張家口 075000)
Gilbert 綜合征(Gilbert syndrome, GS)是一種先天性血清膽紅素增高癥,以黃疸為主要臨床表現。同時合并巨幼細胞貧血非常少見,容易造成漏診。將河北北方學院附屬第一醫院血液科收治的1 例GS 合并巨幼細胞貧血患者報道如下。
1 臨床資料
患者,男性,27 歲。因乏力、食欲不振1 個月,皮膚鞏膜黃染1 周,于2018年7月19日入院。患者原于1 個月前大量飲酒后出現乏力、食欲不振,未引起重視。1 周前發現皮膚鞏膜黃染,在當地醫院查血常規:白細胞3.76×109/L,血紅蛋白92 g/L,平均紅細胞體積105.9 fl,血小板84×109/L,考慮為血液病至本院就診,查血常規:白細胞3.55×109/L,血紅蛋白87 g/L,平均紅細胞體積104 fl,平均紅細胞血紅蛋白量38.3 pg,平均紅細胞血紅蛋白濃度369 g/L,血小板75×109/L,網織紅細胞3.3%;生化指標:谷草轉氨酶47 u/L,總膽紅素79.43 μmol/L,間接膽紅素69.29 μmol/L,乳酸脫氫酶2 025 u/L,α-羥丁酸脫氫酶2 315 u/L。門診以全血細胞減少原因待查收入院。既往史:2年前因貧血、黃疸在當地醫院予以葉酸、維生素B12治療后好轉。查體:體溫36.5℃,脈搏88 次/min,呼吸20 次/min,血壓17.16/7.98 kPa,中度貧血貌,皮膚黃染,無蜘蛛痣及肝掌,鞏膜黃染,心肺無異常,腹平軟,肝脾肋下未觸及,肝區無叩擊痛,無腹水癥,雙下肢無水腫。入院后完善實驗室檢查,血清葉酸濃度:21.49 ng/ml(3.38 ~5.38 ng/ml)、 維 生 素B12濃 度:84 pg/ml(211 ~911 pg/ml);直接及間接抗人球蛋白試驗均陰性;肝炎標志物:乙肝表面抗體為陽性,丙肝抗體為陰性;抗內因子抗體為陰性;外周血涂片:紅細胞大小不等,未見球形紅細胞及紅細胞碎片;骨髓象:骨髓增生極度活躍,粒細胞系占34.5%,紅細胞系占57.5%,紅細胞系增生明顯,以中晚紅為主,部分可見巨幼樣改變,成熟紅細胞大小不等,易見嗜多色紅細胞及點彩紅細胞,全片共見巨核細胞30 個,片中血小板散在(見圖1);胸腹16 排CT 平掃無明顯異常。診斷為巨幼細胞貧血,給予維生素B12治療后,患者癥狀好轉。入院后第10 天復查血常規:白細胞4.9×109/L,血紅蛋白108 g/L,平均紅細胞體積106.2 fl,平均紅細胞血紅蛋白量38.5 pg,平均紅細胞血紅蛋白濃度354 g/L,血小板227×109/L,網織紅細胞7.9%;生化指標:谷草轉氨酶41 u/L,總膽紅素74.43 μmol/L,間接膽紅素61.53 μmol/L,乳酸脫氫酶642 u/L,α-羥丁酸脫氫酶598 u/L。患者黃疸無明顯改善,進一步查紅細胞滲透脆性試驗為陰性;葡萄糖-6-磷酸脫氫酶(glucose-6-phosphate dehydrogenase,G6PD)基因突變檢測為陰性;膽紅素尿苷葡萄糖醛酸轉移酶基因(UGT1A1基因)突變檢測示UGT1A1基因存在變異,位于第1 個外顯子(Exon1)的c.211G>A,最終診斷為GS 合并巨幼細胞貧血。
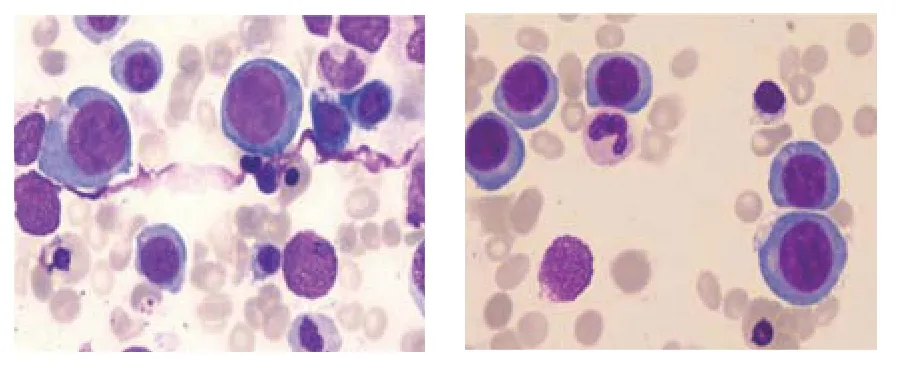
圖1 骨髓象 (光學顯微鏡×400)
2 討論
巨幼細胞貧血是由于葉酸和/或維生素B12缺乏,細胞DNA 合成障礙引起的一種大細胞性貧血。臨床表現為貧血、消化道癥狀、神經系統損害等。因發生骨髓原位溶血,患者常出現間接膽紅素升高的表現。本例患者以乏力、食欲不振、黃疸為主要臨床表現;因工作原因平素飲食不規律;血常規示全血細胞減少,大細胞性貧血;肝功能示膽紅素升高,以間接膽紅素為主;血清維生素B12濃度明顯減低,明確診斷為巨幼細胞貧血,給予維生素B12治療后,貧血逐漸好轉,但膽紅素水平無明顯下降。分析黃疸原因,患者間接膽紅素升高明顯,可排除梗阻性黃疸及肝細胞性黃疸;追問病史,發現患者運動或飲酒后有黃疸表現,且母親及姐姐有間斷黃疸病史,應警惕患者是否存在遺傳性溶血性貧血如G6PD 缺乏癥、遺傳性球形紅細胞增多癥,或遺傳性膽紅素代謝障礙性疾病如GS,因此行紅細胞滲透脆性試驗、G6PD基因突變檢測及UGT1A1基因突變檢測。本例患者外周血涂片未見球形紅細胞,紅細胞滲透脆性試驗為陰性,G6PD基因突變檢測為陰性,可排除遺傳性球形紅細胞增多癥和G6PD 缺乏癥;UGT1A1基因突變檢測到UGT1A1基因存在變異,位于Exon1 的c.211G>A,最終診斷為GS 合并巨幼細胞貧血。
GS 是一種先天性血清膽紅素增高癥,遺傳方式為常染色體隱性遺傳和常染色體顯性遺傳(不完全外顯)。位于染色體2q37 位點的UGT1A1基因發生缺陷,導致尿苷二磷酸葡萄糖醛酸基轉移酶-1(UDPglucuronosyltransferase 1, UGT1)表達下降,間接膽紅素無法轉變為直接膽紅素,因此出現間接膽紅素水平升高。該病多于15 ~30 歲發病,男性多見,多有家族史。起病隱匿,臨床表現為長期間歇性輕度黃疸,患者多無明顯癥狀,少數可能有輕度乏力、納差或肝區不適。劇烈運動、勞累、饑餓、創傷、感染,或者妊娠可能為誘因導致黃疸加重。該病預后一般良好,無需特殊治療,可通過戒酒、休息等方式減輕癥狀,并可試用苯巴比妥誘導UGT 活性,促進膽紅素排泄[1]。該例患者UGT1A1基因的第1 個外顯子第211 位的鳥嘌呤(G)突變為腺嘌呤(A),導致編碼產物中第71位甘氨酸變為精氨酸(Gly71Axg),此突變位點在日本人、韓國人及中國人中常見[2]。
GS 合并巨幼細胞貧血較少見,巨幼細胞貧血可表現為黃疸,以間接膽紅素升高為主,與GS 相混淆,容易造成漏診。因此在臨床中遇到黃疸患者,無法用一種病解釋時,應想到GS。需要臨床醫生仔細詢問病史,完善相關檢查,避免漏診、誤診。
猜你喜歡
英語世界(2023年6期)2023-06-30 06:29:10
中國醫學影像學雜志(2021年6期)2021-08-13 08:43:36
中國生殖健康(2020年2期)2021-01-18 02:51:26
小學生導刊(2018年13期)2018-06-29 03:49:00
中國生殖健康(2018年2期)2018-01-12 13:57:51
現代檢驗醫學雜志(2016年4期)2016-11-15 02:01:14
中國現代醫學雜志(2015年26期)2015-12-23 11:04:22
鄭州大學學報(醫學版)(2015年2期)2015-02-27 14:50:44
中華皮膚科雜志(2014年4期)2014-12-19 12:55:49
中國神經精神疾病雜志(2014年1期)2014-03-01 03:23:22
