制備色譜分離純化桑葉提取物的研究
2020-01-01 09:39:00王露唐付杰徐笑非詹力王星敏
應用化工 2019年12期
王露,唐付杰,徐笑非,詹力,王星敏,2
(1.重慶工商大學 環境與資源學院,重慶 400067;2.重慶市特色農產品加工儲運工程技術研究中心,重慶 400067)
桑葉含有多種生理活性物質,具有降血糖[1]、降血脂[2]、抗炎[3]、抗氧化[4]、抗腫瘤、抗菌抗病毒[5]等多種藥理作用,常作為天然著色劑、抗氧化劑和功能性食品原料[6],國家衛生部將桑葉列為藥食同源的中藥[7],衛計委批準桑葉提取物作為新食品原料[8]。制備型高效液相色譜法(P-HPLC)是目前最成熟、應用最為廣泛的分離純化技術,具有純度高、產率高、分離速度快等優點。目前我國對桑葉開發利用僅為初加工,多為復方制劑,而天然活性成分單體的應用不多,影響桑葉高附加值的提取物經濟推廣。故開展桑葉高活性成分篩選及開發有助于提高桑葉綜合利用,滿足廣大人民群眾醫療健康的需求。
1 實驗部分
1.1 試劑與儀器
桑葉,重慶市農科院;無水乙醇、磷酸均為分析純;乙腈、甲醇均為色譜純;異槲皮苷標準品(≥98%)。
1260高效液相色譜儀;CJF-0.05小型高壓反應釜;DHG-9070A臺式電熱恒溫鼓風干燥箱;SHB-Ⅲ循環水式多用真空泵;KC-100高速粉碎機;IR Prestige-21傅里葉變換紅外光譜儀;KQ-250V超聲波清洗機;Gelailnest半制備色譜儀。
1.2 實驗方法
1.2.1 桑葉粗提液的制備 分別稱取過篩的干燥桑葉5 g于水熱反應釜內膽中,按乙醇體積與桑葉質量比為7∶1 mL/g加入體積分數為57%的乙醇,于154 ℃的烘箱中水熱反應80 min后,過濾,分別收集濾渣和濾液,濾液即含有異槲皮苷醇提液;濾渣集中收集后以待二次利用。
1.2.2 活性成分檢測及分析 用分析型高效液相色譜分析。色譜柱為Hypersil ODS柱(250 mm×4.6 mm,5 μm),流動相是體積比20∶80的乙腈和0.5%磷酸水溶液,流速為1 mL/min,柱溫為25 ℃,進樣量為10 μL;通過對樣品進行分析型液相色譜全掃描,確定異槲皮苷的最大吸收波長為350 nm。
1.2.3 活性成分分離純化 半制備型液相色譜條件為色譜柱(C18-8),流動相為乙腈-水(20∶80);進樣量10 mL,流速20 mL/min;檢測波長350 nm。按照上述方法進Gelailnest制備色譜儀,根據樣品的保留時間進行分段收集,得到4種餾分A、B、C、D。并按照式(1)計算分離度。
(1)
其中,tR2為相鄰兩峰中后一峰的保留時間;tR1為相鄰兩峰中前一峰的保留時間;W1、W2為此相鄰兩峰的峰寬。《中國藥典》規定:R≥1.5,視為完全分離;R<1,部分重疊。通常R=1.5可作為已完全分離的標志。
1.2.4 單體制備 采用冷凍干燥法將收集的 4種餾分和桑葉粗提液分別進行干燥,得到A、B、C、D和提取復合物的粉末狀固體物,稱其質量記為WA、WB、WC、WD和W1。分別準確稱取2 mg,用57%的乙醇溶解,定容于25 mL容量瓶中,采用高效液相色譜分析方法進行純度測定,代入標準曲線計算出濃度C,則制備率的計算公式如下:
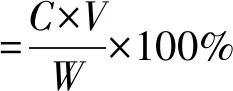
(2)
式中C——粗提液中各成分的濃度,mg/mL;
V——粗提液的體積,mL;
W——制備得到的各單體的量,mg。
1.2.5 產品紅外分析 采用衰減全反射模式測定制備得到的4種成分的傅里葉變換紅外光譜,分析制備得到的粉末的表面官能團,測定波數為4 000~500 cm-1。
2 結果與討論
2.1 分離純化適宜條件篩選
2.1.1 流動相體系及配比的選擇 當流速為20 mL/min,進樣量為10 mL,分別考察了不同比例的甲醇-水和乙腈-水為流動相對異槲皮苷分離效果的影響,結果見表1。

表1 不同流動相體系及配比對異槲皮苷分離效果的影響
由表1可知,隨著溶劑極性的減小,異槲皮苷的保留時間、分離度及異槲皮苷質量分數逐漸減小。當甲醇的比例低于30%時,流動相洗脫能力不強,異槲皮苷不能和其它活性成分很好的分離,并且拖尾嚴重,導致異槲皮苷收集不完全,從而使得產品的純度較低;當甲醇比例大于50%時,雖能在較短時間得到組分,但由于洗脫能力過強,使得異槲皮苷與其他成分無法分離[9],純度低。而當流動相為V乙腈∶V水=20∶80時,分離度為1.74,分離效果好,其純度為98.13%。與甲醇體系洗脫比較,異槲皮苷在甲醇體系中靠近異槲皮苷的其它活性成分為黃酮類物質,故比較難分開,而乙腈則有一定的手性拆分能力[10],所以乙腈體系可以得到較高質量分數的異槲皮苷及其它成分。
2.1.2 流速對異槲皮苷分離效果的影響 當V乙腈∶V水=20∶80,進樣量為10 mL時,分別考察了流速為15,20,25 mL/min異槲皮苷分離效果的影響,結果見圖1。
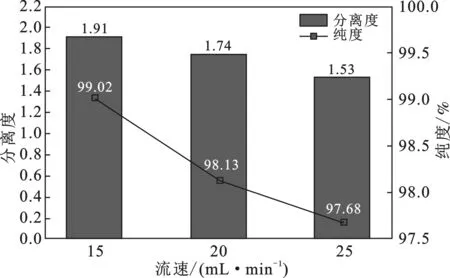
圖1 流速對異槲皮苷分離效果的影響
由圖1可知,隨著流速的增大,異槲皮苷的保留時間減少,分離度降低,樣品質量分數減小。較低的流速一般峰高較高,分離度也較大,但流速低,洗脫時間延長[11]。若以15 mL/min洗脫時,樣品的保留時間較長,流動相用量多,蒸餾費時。而以25 mL/min洗脫時,流速增加,因固定相內擴散阻力起控制作用,溶質組分在固定相和流動相兩相之間的分配偏離分配平衡的幅度增加,分離度降低,色譜峰擴展加劇,因而溶質稀釋程度增加,流動相消耗增大,同時也增加了經濟負擔[12]。綜合各種因素,實驗選擇20 mL/min的流速進行考察。
2.1.3 粗提液不同進樣量對異槲皮苷分離效果的影響 當V乙腈∶V水=20∶80,流速為20 mL/min時,考察了進樣量分別為5,8,10 mL對異槲皮苷分離效果的影響,結果見圖2。通過增加進樣量可以提高制備率[13],但分離度卻隨著進樣量的增加呈現下降趨勢。
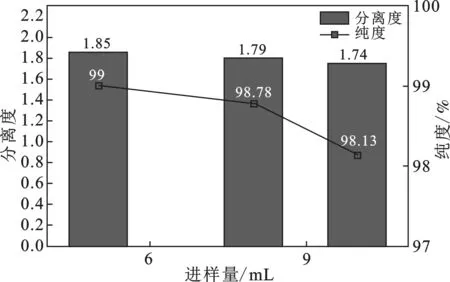
圖2 進樣量對異槲皮苷分離效果的影響
由圖2可知,異槲皮苷的分離度隨著進樣量增大而降低,但是仍然大于1.5,可視為完全分離,異槲皮苷質量分數98.78%,在保證純度的情況下,盡量增加樣品處理量,提高制備率,因此,進樣量為10 mL 最為合適。
2.2 產品檢測與分析
2.2.1 制備單體的HPLC定性分析 將1.2.4節配制好的制備單體溶液按HPLC分析條件進樣分析,與標品溶液出峰時間進行對照,具體情況見表2,可以確定A、B、C、D 分別是綠原酸、蘆丁、異槲皮苷和紫云英苷。因此,通過單次PHPLC分離除了可以得到異槲皮苷,還可得到綠原酸、蘆丁和紫云英苷3種活性物質,整個過程用時較短、毒性低、分離效果較好,可連續進樣制備,桑葉提取液的半制備液相色譜圖出峰情況見圖3。
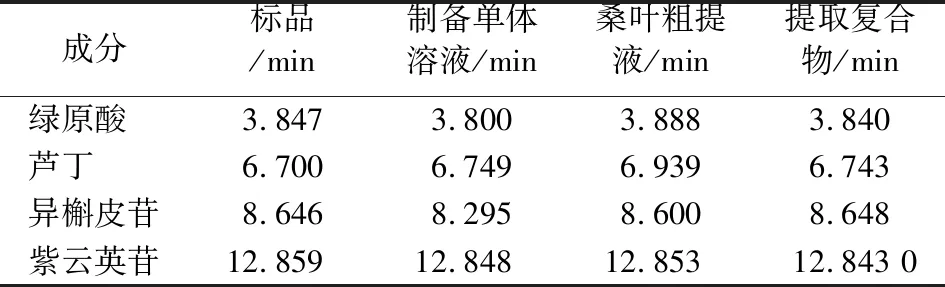
表2 制備單體溶液與標品溶液高效液相出峰時間對照表
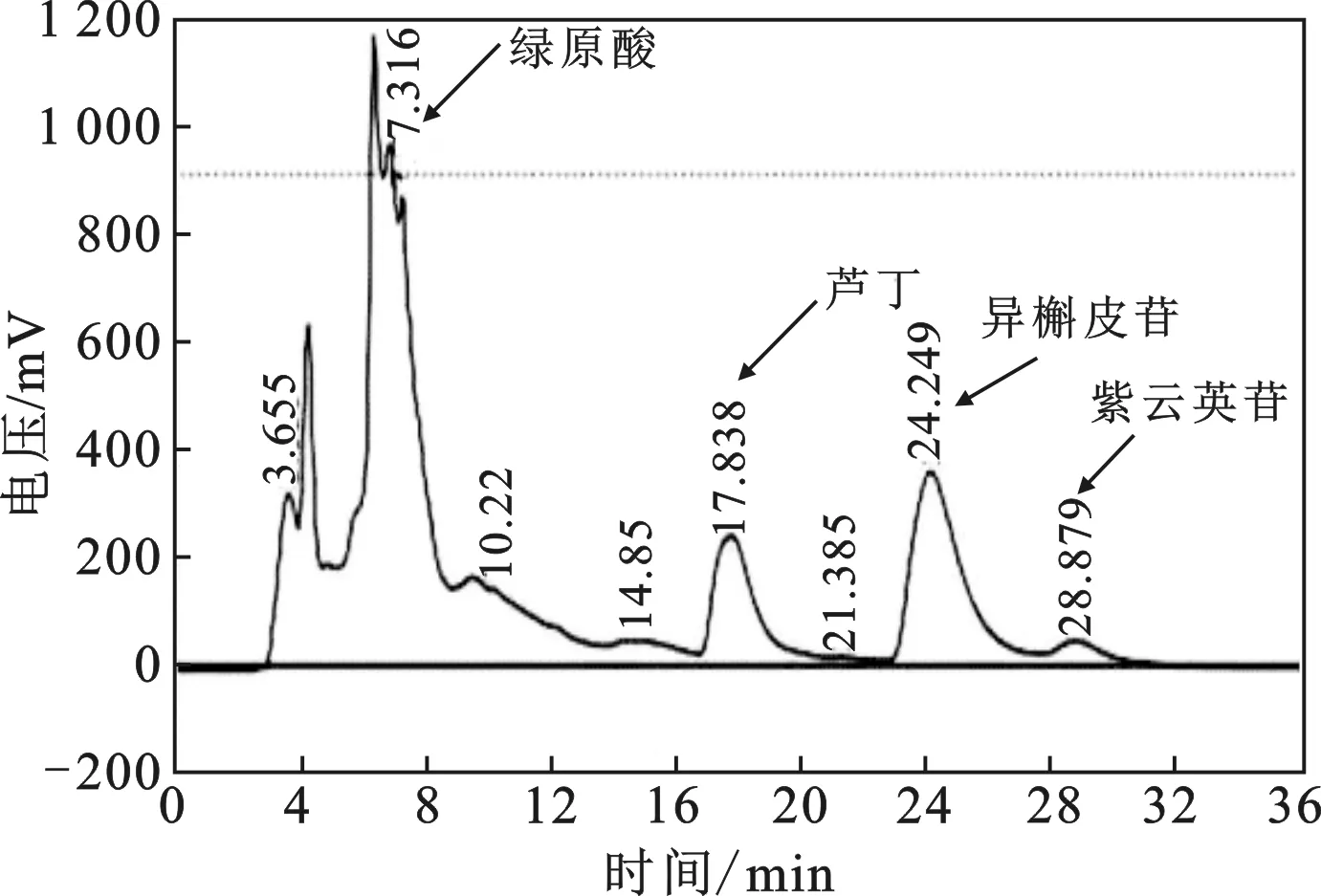
圖3 粗提物溶液的半制備液相色譜圖
2.2.2 制備單體的紅外分析 通過測定制備單體的紅外光譜,結果見圖4。
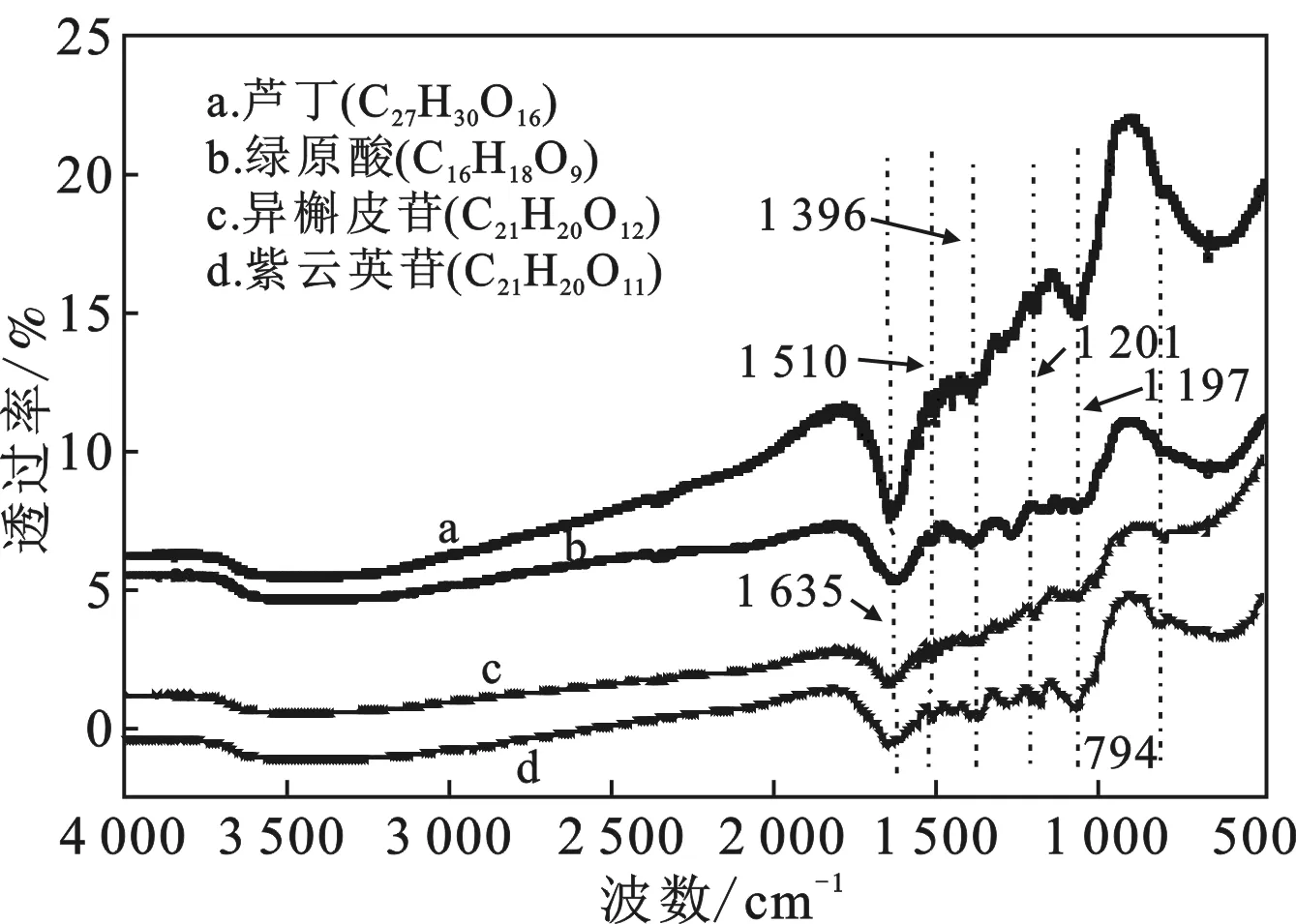
圖4 紅外光譜圖Fig.4 The infrared spectrogram

2.3 制備率計算及效益分析
2.3.1 制備單體的制備率計算 合并多次粗提液共500 mL,其中綠原酸、蘆丁、異槲皮苷和紫云英苷的濃度分別為0.501 8,0.157 8,0.369 4,0.068 mg/mL,可得出500 mL溶液中含有的綠原酸、蘆丁、異槲皮苷和紫云英苷的量分別是250.9,78.9,182.45,34 mg。將500 mL粗體液濃縮至20 mL后,半制備色譜連續進樣2次,收集合并相應餾分,經冷凍干燥后稱其質量W,按照式(2)計算各組分的得率,結果見表3。
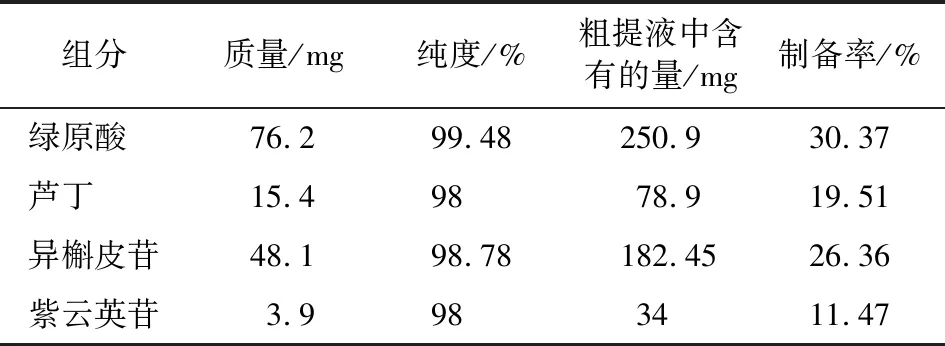
表3 制備單體的制備率
2.3.2 效益分析 除了目標產物的質量分數、產量、消耗時間,成本也是PHPLC的主要考慮因素。市面上,乙腈(色譜純)售價360元/4 L,即乙腈為0.09元/mL,按照V乙腈∶V水=20∶80為流動相20 mL/min為流速時,單次制備需要耗時40 min,乙腈的用量是160 mL,即消耗的流動相的成本為14.4元/次。國內市場上含量≥98%的異槲皮苷售價為1 000元/20 mg,含量≥98%的綠原酸售價為139元/20 mg,含量≥98%的蘆丁售價為140元/20 mg,含量≥98%的紫云英苷售價為500元/20 mg。本次實驗所得到的4種產品的質量根據市面價格折算,預計收益約3 091元。
3 結論
(1)乙腈作為流動相分離效果較甲醇更好,最終適宜的分離純化條件為:V乙腈∶V水=20∶80,進樣量10 mL,流速20 mL/min。
(2)通過單次PHPLC分離除了可以得到異槲皮苷,還可得到綠原酸、蘆丁和紫云英苷3種活性物質,純度均≥98%,制備率分別為26.36%,30.37%,19.51%,11.47%。
(3)每進樣制備一次所消耗的乙腈成本為14.4元 ,本實驗制備單體的量按照市面標準品的價格來折算,效益預測約為3 091元。
