腦神經化學活體原位電化學分析研究進展
2019-11-12 06:29:25薛亦飛肖通方蔣亞楠吳菲于萍毛蘭群
分析化學 2019年10期
薛亦飛 肖通方 蔣亞楠 吳菲 于萍 毛蘭群

摘?要?腦科學已經成為多學科交叉研究的前沿領域之一, 其中腦神經化學的研究由于能夠揭示腦活動和腦疾病過程中的物質基礎, 在神經科學和化學等領域引起了高度關注。電化學分析方法具有高靈敏度、高時空分辨、電極/溶液界面可設計等特點, 尤其適用于在活體動物層次開展腦神經化學的分析研究。本文圍繞活體原位電化學分析方法的原理和特點, 綜述了近年來電化學分析方法在腦神經化學研究中的應用, 并對其未來的發展前景進行了展望。
關鍵詞?活體電化學分析; 腦神經化學; 原位檢測; 評述
1?引 言
腦科學是目前國際前沿科技的熱點研究領域之一, 對腦功能的研究有助于理解人類認知、情感等復雜生理過程的本質, 以及神經系統疾病的形成和發展規律。腦神經信號的傳遞以及代謝過程都離不開化學物質的參與, 因此, 針對腦內神經遞質、調質、能量代謝物質、自由基、離子等諸多神經化學物質開展腦神經分析化學研究, 對于探索和認識神經生理、病理的分子機制, 都具有極其重要的意義。
腦神經化學物質的分析一般分為單囊泡、單細胞、腦片和活體等不同層次。其中, 在單囊泡、單細胞及腦片層次上進行化學物質檢測脫離了活體生存的真實環境, 較難保持細胞之間固有的聯系和相互作用。相比較而言, 活體層次對腦化學物質進行分析, 能夠更加真實、直接地反映神經系統在各種生理、病理過程中對外界刺激的響應, 因而能夠為腦神經生理、病理過程物質基礎的探索提供最為直接的信息。
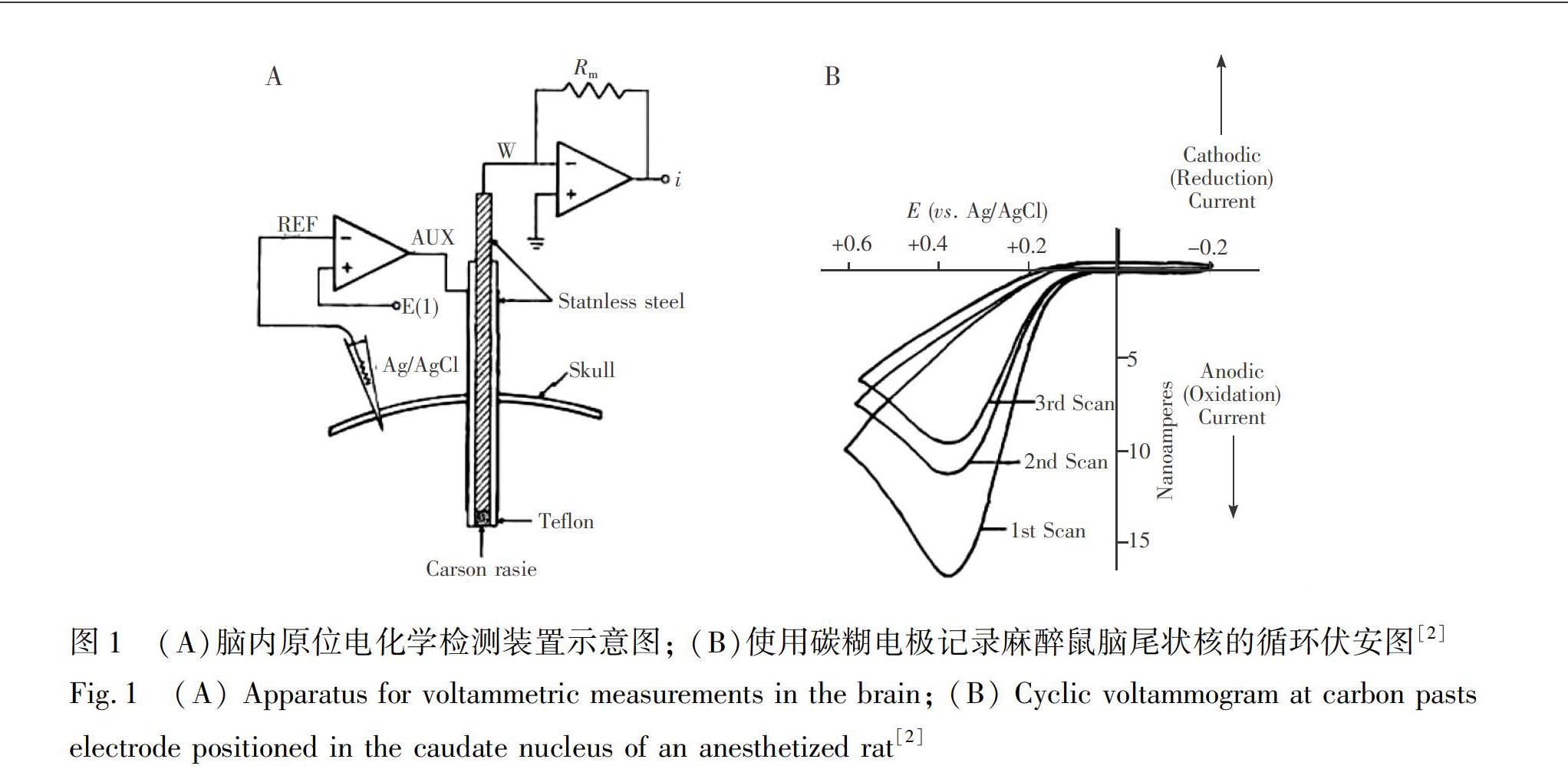
電化學分析方法通常具有靈敏度高、選擇性好、時空分辨率高等優點, 且檢測電極易于微型化, 適用于活體原位分析測定。活體原位電化學分析方法可望應用于腦內不同化學物質基礎水平及其在一系列生理、病理過程中濃度變化的監測。腦神經活體原位電化學分析可追溯到20世紀50年代, Clark等[1]利用玻璃封裝的鉑絲作為研究電極, 首次通過電化學伏安法實現了腦內氧氣濃度變化的實時監測。但是, 這一研究并沒有引起研究者們的廣泛關注。更為人們熟知的是, 1973年Adams 等[2]首次將微型碳糊電極植入大鼠腦中進行活體電化學研究, 其電極結構如圖1A所示。該研究得到了活體腦內的第一張循環伏安圖(圖1B),進一步驗證了在腦內使用電化學方法實現生理活性物質檢測的可行性, 引起了神經生理學家的高度關注, 標志著活體原位腦神經電化學分析的誕生。
近年來, 隨著分析科學、化學、電子科學、神經科學等多學科的快速發展和交叉融合, 腦神經活體原位電化學分析也不斷發展完善, 為相關生理、病理過程研究提供了重要的實驗方法, 進一步推動了分析化學與腦神經科學的實質性交叉與融合。本文著重介紹活體原位電分析化學方法的原理、特點及其在腦神經化學研究中應用的進展, 并對其發展趨勢進行展望。
2?活體原位電化學分析方法
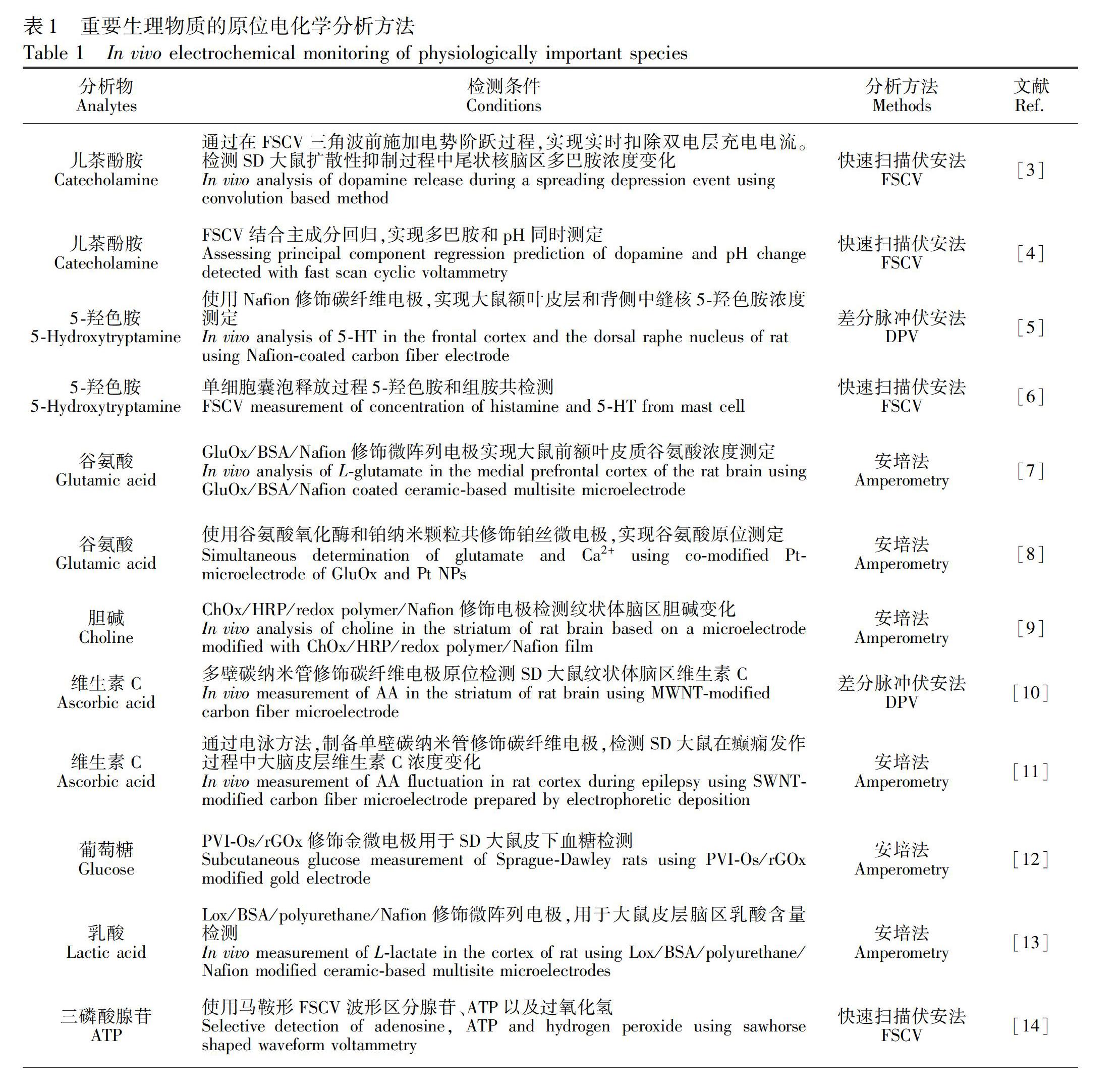
目前,在神經科學領域, 利用電化學分析方法可實現多種生理活性物質的活體原位實時分析。表1列舉了一些重要的生理活性物質及其活體原位電化學分析方法。
2.1?基于伏安法的活體原位電化學分析
伏安法是一類重要的電化學測量方法, 通過向電極施加調制的電壓波形, 測量電化學體系的電流響應, 從而獲得電極過程的電位?電流關系。通過對伏安曲線波形和峰高等參數進行分析, 可實現對于具有不同電化學參數的電化學活性物質進行定性與定量分析。伏安法選擇性高, 可實現單一或多種物質同時的選擇性分析。然而, 伏安法的時間分辨率通常會受到掃描速率的限制, 同時施加的調制電壓波形也會對分析體系產生一定影響。
根據伏安法檢測過程中施加波形的不同又可將其分為脈沖伏安法和電勢掃描伏安法。其中, 脈沖伏安法的特點是可有效抑制背景充電電流, 并降低擴散層變化的影響, 具有靈敏度高、選擇性高、可同時區分多種電化學活性物質等優勢, 但時間分辨率比較低, 無法記錄快速變化過程。自20世紀70年代以來, 以差分脈沖伏安法(Differential pulse voltammetry, DPV)為代表, 并包括常規脈沖伏安法(Normalpulse voltammetry, NPV)、 差分常規脈沖伏安法(Differential normal pulse voltammetry, DNPV)及方波伏安法(Square wave voltammetry, SWV)等在內的脈沖伏安法逐漸被應用于腦內多種神經遞質的同時檢測。為了提高伏安法的時間分辨率, 實現短時間遞質快速變化過程的實時分析, 以快速掃描循環伏安法(Fast scan cyclic voltammetry, FSCV)為主的電勢掃描伏安法在近幾十年中得到了很好的發展。FSCV方法可實現快速分析, 但背景電流大, 難以用于神經化學物質基礎水平的檢測和長時程記錄。目前, 該方法主要應用于多巴胺刺激釋放等電化學活性神經化學物質的快速變化過程研究。
2.1.1?DPV方法?DPV法是一種脈沖伏安測量技術。檢測過程中, 直流電壓以一定頻率和幅值進行步進掃描, 并在每次步進時疊加固定幅值(10~100 mV)的方波脈沖, 記錄脈沖結束前和脈沖開始前的電流差值。DPV檢測過程中采用的特殊脈沖波形、采樣方法以及電流差減運算配合原位電化學檢測使用的微電極, 可有效排除雙電層充電電流以及擴散層變化的影響, 顯著提高分析方法靈敏度和選擇性。對于氧化電位差大于100 mV的電活性物質, 使用DPV方法可有效排除氧化還原電流的相互干擾, 實現不同物質的區分及定量分析。
DPV方法在活體原位電化學分析領域的應用可追溯到1976年, Lane等[22]首次使用經KI溶液處理過的鉑微電極作為研究電極, 通過DPV方法實現了Sprague?Dawley(SD)大鼠尾狀核維生素C和兒茶酚胺濃度的同時檢測, 其電極結構如圖2A所示。隨后, Gonon等[23]使用DPV方法實現了麻醉大鼠紋狀體腦區電化學活性神經化學物質的原位分析, 記錄到的DPV曲線如圖2B所示。通過干預實驗對比, 最終將記錄到的兒茶酚電流信號歸屬為DOPAC。此后, DPV方法逐漸被應用于電活性神經化學物質的多組分同時分析[24~26]。
DPV方法不僅可實現具有電化學活性神經化學物質的直接分析, 也可利用非電化學活性物質與電極修飾材料間的相互作用, 實現部分電化學活性較差的神經分子的間接檢測。Chai等[27]使用無銅衍生的超氧化物歧化酶(E2Zn2SOD)作為生物識別元件, 利用其與Cu2+的特異性相互作用, 構筑了用于Cu2+檢測的電極界面, 利用DPV法實現了活體原位Cu2+的檢測, 方法靈敏、可靠。Zhao等[28]合成了一種二茂鐵吡啶衍生物(N?(6?aminopyridin?2?yl)ferrocene, Fc?Py), 利用吡啶作為質子響應單元, 二茂鐵作為電化學響應單元, 通過DPV方法實現了pH值的活體原位分析。
2.1.2?FSCV?FSCV是一種具有高時間分辨率的電勢掃描伏安法。以碳纖維微電極作為研究電極, 并以一定頻率施加大于100 V/s 掃速的三角波進行循環伏安分析, 可達到毫秒級的時間分辨率[29]。在FSCV分析中, 隨著電位掃描速度的增加, 電化學反應動力學較慢的物質將表現得更不可逆, 其氧化還原峰偏移程度大于電化學反應速度較快的電活性物質, 從而實現對不同電極過程動力學的物質進行區分。FSCV與常規掃速(1~500 mV/s)下的循環伏安法不同, 電位掃描過程中, 由于雙電層充放電電流與掃速成正比, 而法拉第電流與掃速的平方根成正比。因此,高掃速下,循環伏安曲線中雙電層充放電所貢獻的非法拉第電流會明顯增大, 并成為能記錄到的總電流的主要組成部分, 影響對目標分析物氧化還原過程法拉第電流的提取。通常,FSCV的結果會以變化前某一特定周期的FSCV數據作為背景進行扣除, 以便降低背景電流的影響[30]。扣除背景后的FSCV結果可進行定性和定量分析。但是,由于背景扣除過程所扣除的一般是固定的循環伏安數據, 這將導致在長時間電化學分析中雙電層結構變化所造成的背景電流漂移不斷積累, 對分析結果產生較大影響。因此, 該方法持續分析時間通常約1 min, 不宜進行長時間的連續分析。此外, 背景扣除導致了FSCV法很難實現腦內神經化學物質的基礎水平測定[31]。
目前, FSCV方法應用較為成熟的測定體系是對腦內兒茶酚胺類神經遞質(尤其是多巴胺)快速變化過程的原位實時分析[32]。活體分析過程中, 在每個施加的三角波之間通常會將研究電極維持在負電位(如Symbolm@@
0.4 V)下, 利于兒茶酚胺類物質在電極表面的富集, 從而提高檢測的靈敏度[33]。同時, 帶負電的物質不會得到富集,在一定程度上也提高了FSCV對兒茶酚胺類物質檢測的選擇性。在高掃速下, 多巴胺等具有較快電子轉移速率的電化學活性物質的氧化還原電流得到大幅提高; 另一方面也使得抗壞血酸等電子轉移速度相對較慢的干擾物質在電極上表現得更不可逆, 其氧化峰正移, 進而實現對多巴胺等物質的選擇性檢測[34,35]。
FSCV方法雖然具有良好的選擇性和靈敏度, 并能夠實現高時間分辨, 但由于腦神經系統中多種電化學活性生理物質具有相近的氧化電位, 導致產生的氧化還原電流相互交疊, 不容易區分與精準定量。因此, 早期的FSCV方法主要用作神經化學過程中電化學活性物質的定性和半定量研究。Wightman 等[36]利用不同顏色代表不同電流值, 將一組FSCV數據拼合繪制成“電位?時間?電流”二維圖, 即可通過二維圖中不同的圖案直觀地看出不同時間不同電化學活性物質濃度的變化過程, 為人們研究復雜生理病理過程中物質的變化提供了可能。
采用FSCV方法進行定量分析時, 通常在目標分析物FSCV結果中選取氧化還原電流較大, 且無其它物質干擾電位下的電流值, 以此繪制出電流隨時間變化的曲線, 或根據線性關系換算為濃度?時間曲線。然而, 這種方法在多種物質變化的復雜體系中存在很大的局限性。為了解決FSCV方法在復雜體系中的定量問題, Heien等[37]將主成分回歸(Principal component regression, PCR)應用于多種物質變化的復雜體系下FSCV結果的定量分析。他們發現, 多巴胺的FSCV波形與不同pH值造成的FSCV波形變化有明顯差異, 如圖3A和3B所示。采用主成分回歸方法可將FSCV波形中多巴胺的氧化還原波形和pH值造成的波形變化進行有效地區分, 并逐漸發展出多巴胺和pH值雙組分同時測定的方法[4, 38]。隨后, Heien等[39]利用該方法研究了可卡因對尾狀核腦區多巴胺釋放的調控, 以及該過程中的pH值變化(圖3C)。
在FSCV分析時, 主成分回歸方法雖然可用于解決多種神經化學物質的區分和定量問題, 但由于雙電層變化導致的背景電流漂移卻無法通過主成分回歸消除。為了使主成分回歸算法計算結果真實有效, FSCV方法應用于活體分析的持續時間通常短于90 s[4]。很多研究組也通過優化FSCV分析中所使用的電極和數據處理方法, 使其能夠進行長時間的活體分析。如Clark等[40]將直徑7 μm的碳纖維封裝在直徑90 μm的熔融玻璃毛細管中, 發現電極尺寸的減小可明顯降低其對腦組織的損傷, 并減少免疫反應, 從而使電極具有良好的生物兼容性。利用此電極, 實現了對腦內多巴胺長達10~16 周的活體分析。Schwerdt等[41]進一步優化了長時間記錄時FSCV的數據處理方法, 通過將持續記錄的FSCV數據分割成多個50 s時長, 分別進行主成分回歸分析, 再將分析結果拼合即可得到較長時間的檢測結果, 最終實現了使用長期植入陣列電極進行非人靈長類動物腦內多巴胺變化的長時程監測。
2.2?基于安培法的活體原位電化學分析
安培法(Amperometry)是一種通過向研究電極施加恒定電位, 從而實現對電化學活性物質定量分析的電化學方法。其特點在于, 測量過程中研究電極維持恒定電位, 從而降低雙電層充電電流的影響。因此, 相對于FSCV方法, 安培法可進行長時間的持續分析檢測。但是, 安培法只能提供某一固定電位下的電流信息, 因此, 分析方法的選擇性完全取決于電極界面的反應和設定的電位。近年來, 安培法逐漸成為活體原位電化學分析中的主要方法之一。通過合理構筑電極/溶液界面, 調控物種的電極反應動力學, 一些神經化學物質(如維生素C、O2、H2O2、NO、H2S)都可通過安培法實現活體原位分析。
2.2.1?維生素C的活體原位電化學分析?維生素C(Vitamin C or ascorbic acid, AA)是神經系統中的一種重要神經化學物質, 具有較強的還原性, 在許多生理病理過程中發揮著重要的作用。在多數碳電極表面, 維生素C的電化學過程表現為內殼層反應, 極易受到電極表面物理化學性質的影響。同時, 維生素C的電化學氧化會產生具有非電化學活性、易吸附于電極表面的水解產物, 導致電極靈敏度下降。因此, 維生素C在常規的碳纖維電極上表現出較慢的電子轉移速率、高的過電位和較差的穩定性[42, 43]。Zhang 等[10, 44]首先發現在碳納米管修飾電極上維生素C具有較低的氧化過電位, 可用于維生素C的原位檢測(圖4A)。他們使用多壁碳納米管修飾碳纖維電極在維生素C溶液中進行循環伏安分析, 實驗結果表明, 維生素C的氧化電流在0.0 V(相比于標準Ag/AgCl電極)就達到了穩態(圖4B)。該電位下不會發生多巴胺、5?羥色胺、DOPAC等其它腦內常見神經化學物質的氧化, 說明多壁碳納米管修飾碳纖維電極可實現維生素C的高選擇性分析(圖4C)。此外, 他們發現碳納米管電極還可提高維生素C連續測定的穩定性。基于這些性能, 成功建立了鼠腦內維生素C的活體原位電化學分析新方法。
隨后, Xiang等[45]制備了陣列碳納米管包覆的碳纖維電極, 將電極植入鼠腦紋狀體, 并施加+0.05 V(vs. Ag/AgCl)的極化電壓, 利用安培法實現了腦內維生素C的活體原位分析。基于該方法高的時空分辨率, 通過在微電極附近灌注谷氨酸溶液, 他們在活體層次觀察到了腦神經系統內抗壞血酸?谷氨酸的“異相交換”過程。
Xiao等[11,46]利用電泳的方法將單壁碳納米管沉積于碳纖維電極的表面, 并通過高溫和電化學處理, 得到了對維生素C具有高選擇性和高穩定性的碳納米管電極。該方法制備的電極具有高度的一致性, 解決了碳納米管在碳纖維電極表面制備重現性差的問題。他們將制備的電極植入鼠腦皮層, 在+0.05 V極化電位下, 首次在活體動物層次觀測到了癲癇模型和擴散性抑制過程中腦內維生素C濃度升高的現象(圖5A和5B), 為進一步研究這些病理過程中的分子機制提供了直接的實驗基礎。
2.2.2?氧氣的原位電化學檢測?氧氣在生命體中發揮著重要的作用。氧氣供給不足會造成神經系統能量代謝困難, 增加氧化應激壓力, 甚至造成細胞損傷。因此, 對腦內氧氣濃度的原位檢測將直接反映神經系統的活動功能。利用安培法進行氧氣活體原位分析的關鍵問題在于, 在以金和碳等材料為基底的電極表面, 氧氣電化學還原過程通常以兩步兩電子還原為主, 生成過氧化氫中間產物[47]。而在鉑基電極材料表面, 氧氣電化學還原過程以四電子還原為主, 最終生成水[48]。在活體原位分析中, 考慮到氧氣兩電子電化學還原過程生成的過氧化氫中間產物可能會對神經系統造成損傷[49], 所以鉑基電極能夠較好適用于腦內氧氣的活體電化學分析[50, 51]。Xiang 等[52]使用電沉積的方法, 在垂直生長碳納米管的碳纖維電極(圖6A)上通過循環伏安法電化學沉積鉑, 最終得到表面均勻覆蓋鉑納米顆粒的修飾電極(圖6B)。在Symbolm@@
0.5 V的電位下, 該電極對氧氣檢測表現出較高的靈敏度和選擇性, 其它腦內常見的生理活性物質均不會對氧氣檢測產生干擾(圖6C)。為了進一步降低電極表面蛋白污染, 提高電極對氧氣檢測的穩定性, 他們用Nafion膜修飾電極, 最終實現了氧氣的四電子電化學還原和選擇性測定, 以及大鼠海馬腦區在全腦缺血?再灌注時氧氣濃度變化的活體原位分析(圖6D)。
2.2.3?基于離子傳輸的原位電化學分析?神經生理物質中的非電化學活性分子, 一般可采用生物傳感器進行檢測。然而酶等生物識別元件有限, 限制了該方法在活體原位電化學分析中的發展。近年來, 基于離子傳輸構建的電化學分析方法逐漸成為活體分析新的發展方向[53]。He等[54]首次報道了聚電解質刷修飾后玻璃微米管的整流行為。在此基礎上, Zhang 等[55]通過在玻璃微米管內壁共修飾聚電解質刷以及Aptamer實現了ATP的選擇性檢測。 Colombo等[56, 57]通過在玻璃納米孔中構建液?液界面, 基于離子傳輸原理實現了對多巴胺、乙酰膽堿、5?羥色胺的檢測。由于這一電化學分析新方法的原理是對微/納孔道內離子流進行檢測, 不需要待測物在電極表面發生電化學氧化還原反應, 預期該方法在電化學惰性物質的活體電化學分析中具有很高的應用潛力。
2.3?基于電位法的活體原位電化學分析
電位法(Potentiometry)是一種在開路狀態下, 直接記錄研究電極相對于參比電極的電位來分析電極表面目標物濃度的分析方法。相對于伏安法和安培法, 電位法的優勢在于, 測量回路中電流幾乎為零, 排除了因測量而產生的電流對腦神經的影響。根據能斯特方程, 在平衡狀態下, 電極開路電位(Open?circuit potential, OCP)的變化是由電極溶液界面各種化學物質的活度決定的。通過設計選擇性識別單元, 可顯著提高開路電位對特定物質濃度變化響應的靈敏度, 并降低其它物質的干擾, 從而實現定量分析。
電位法通常用于離子等非電化學活性物質測定, 例如, 在微電極表面修飾離子選擇性膜作為識別單元, 構建全固態離子選擇性電極, 可實現腦內H+、K+、Ca2+等活體原位分析。Hao等[58]將質子選擇性膜修飾于碳纖維電極上, 制備了對H+具有選擇性好和抗污染性能強的全固態pH選擇性電極, 電極靈敏度達到58.4 mV/pH, 接近Nernst方程的理論值。利用該電極, 他們原位檢測了大鼠吸入CO2后恐慌過程中腦內pH值變化。
除了對離子進行檢測, 近年出現了一種基于原電池原理的電位測定方法(Galvanic redox potentiometry, GRP), 利用陽極作為指示電極測定分析物電化學氧化電位, 陰極作為參比電極, 通常設計為氧還原過程。使用具有高電阻的電位計將陰極、陽極連成回路(圖7A)。由于回路具有高阻抗, 因此回路中電流很小, 電極過程近似處于熱力學平衡態。當陰極還原電位高于陽極氧化電位時, 整個回路中的電化學過程可自發進行, 其輸出的電壓值可作為定量物質濃度的指標[59], 最終實現電化學活性物質的檢測分析。Wu等[60]利用漆酶修飾的碳纖維電極插入到毛細管內指示溶液中氧氣還原電位, 提高了該GRP體系中作為參比的陰極氧氣的還原電位。檢測過程中毛細管內溶液氧氣濃度保持恒定, 提高了體系的靈敏度和穩定性。蛋白吸附污染并不會影響GRP體系的靈敏度(圖7B), 說明該方法具有較高的抗污染干擾能力。他們將碳納米管修飾的碳纖維電極陽極與漆酶修飾的碳纖維電極陰極植入大鼠的大腦皮層, 實現了大鼠全腦缺血/再灌注過程中腦內抗壞血酸的活體原位測定(圖7C)。
3?總結與展望
腦神經活體原位電化學分析是活體分析研究的重要手段之一, 不同電化學方法根據其特點和優勢可用于不同的活體原位分析。目前, 通過選擇合適的電化學方法, 設計合理的電極界面, 可實現多種神經生理物質的原位分析, 對于腦科學的研究起到了較好的推動作用。然而, 目前仍然還有很多重要神經生理物質, 尤其是非電化學活性物質, 仍然難以通過現有的電化學分析方法進行活體原位檢測。而許多已有的活體原位電化學分析方法, 仍然面臨著問題和挑戰。例如, 利用微電極進行活體原位分析檢測時, 普遍存在電極在活體環境中的穩定性和抗污染問題, 活體檢測環境與體外校正環境存在差異等。對于活體原位電化學分析研究而言, 其最終目標是, 在最大程度減少對實驗動物損傷影響的條件下, 精準地記錄生理和病理過程中神經分子的變化。因此, 電極/腦界面的設計和調控[61,62]、電極表面抗污染[63, 64], 電極進一步微型化[65,66]、柔性化[67]、記錄設備小型化、無線化[68~70]等, 都應為未來的重點研究內容。此外, 利用所發展的活體原位電化學分析方法, 實現與腦神經科學家的實質性合作, ?在活體層次發現并描述腦神經生理和病理過程的分子事件, 也將是活體電化學分析領域的研究核心之一。
References
1?Clark L C, Misrahy G, Fox R P. J. Appl. Physiol., 1958, 13(1): 85-91
2?Kissinger P T, Hart J B, Adams R N. Brain Res., ?1973, ?55(1): 209-213
3?Johnson J A, Hobbs C N, Wightman R M. Anal. Chem., ?2017, ?89(11): 6166-6174
4?Keithley R B, Wightman R M. ACS Chem. Neurosci., ?2011, ?2(9): 514-525
5?Crespi F, Martin K, Marsden C. Neuroscience, ?1988, ?27(3): 885-896
6?Travis E R, Wang Y M, Michael D J, Caron M G, Wightman R M. Proc. Natl. Acad. Sci. USA, ?2000, ?97(1): 162-167
7?Burmeister J J, Gerhardt G A. Anal. Chem., ?2001, ?73(5): 1037-1042
8?ZHAO Fan, SHI Guo?Yue, TIAN Yang. Chinese J. Anal. Chem., ?2019, ?47(3): 347-354
趙 凡, 施國躍, 田 陽. 分析化學, 2019, ?47(3): 347-354
9?Garguilo M G, Michael A C. J. Am. Chem. Soc., ?1993, ?115(25): 12218-12219
10?Zhang M, Liu K, Xiang L, Lin Y, Su L, Mao L. Anal. Chem., ?2007, ?79(17): 6559-6565
11?Xiao T, Jiang Y, Ji W, Mao L. Anal. Chem., ?2018, ?90(7): 4840-4846
12?Csoeregi E, Quinn C P, Schmidtke D W, Lindquist S?E, Pishko M V, Ye L, Katakis I, Hubbell J A, Heller A. Anal. Chem., ?1994, ?66(19): 3131-3138
13?Burmeister J J, Palmer M, Gerhardt G A. Biosens. Bioelectron., ?2005, ?20(9): 1772-1779
36?Michael D, Travis E R, Wightman R M. Anal. Chem., ?1998, ?70(17): 586A-592A
37?Heien M, Johnson M A, Wightman R M. Anal. Chem., ?2004, ?76(19): 5697-5704
38?Keithley R B, Heien M L, Wightman R M. TrAC?Trends Anal. Chem., ?2009, ?28(9): 1127-1136
39?Heien M, Khan A S, Ariansen J L, Cheer J F, Phillips P E M, Wassum K M, Wightman R M. Proc. Natl. Acad. Sci. USA, ?2005, ?102(29): 10023-10028
40?Clark J J, Sandberg S G, Wanat M J, Gan J O, Horne E A, Hart A S, Akers C A, Parker J G, Willuhn I, Martinez V, Evans S B, Stella N, Phillips P E M. Nat. Methods, ?2010, ?7(2): 126-132
41?Schwerdt H N, Shimazu H, Amemori K I, Amemori S, Tierney P L, Gibson D J, Hong S, Yoshida T, Langer R, Cima M J, Graybiel A M. Proc. Natl. Acad. Sci. USA, ?2017, ?114(50): 13260-13265
42?Pisoschi A M, Pop A, Serban A I, Fafaneata C. Electrochim. Acta, ?2014, ?121: 443-460
43?Cheng H, Li L, Zhang M, Jiang Y, Yu P, Ma F, Mao L. TrAC?Trends Anal. Chem., ?2018, ?109: 247-259
44?Zhang M, Liu K, Gong K, Su L, Chen Y, Mao L. Anal. Chem., ?2005, ?77(19): 6234-6242
45?Xiang L, Yu P, Hao J, Zhang M, Zhu L, Dai L, Mao L. Anal. Chem., ?2014, ?86(8): 3909-3914
46?Xiao T, Wang Y, Wei H, Yu P, Jiang Y, Mao L. Angew. Chem. Int. Edit., ?2019, ?58(20): 6616-6619
47?Kruusenberg I, Alexeyeva N, Tammeveski K. Carbon, ?2009, ?47(3): 651-658
48?Chen Z, Waje M, Li W, Yan Y. Angew. Chem. Int. Edit., ?2007, ?46(22): 4060-4063
49?Ledo A, Lourenco C F, Laranjinha J, Brett C M, Gerhardt G A, Barbosa R M. Anal. Chem., ?2017, ?89(3): 1674-1683
50?Jung S K, Gorski W, Aspinwall C A, Kauri L M, Kennedy R T. Anal. Chem., ?1999, ?71(17): 3642-3649
51?You T, Niwa O, Tomita M, Hirono S. Anal. Chem., ?2003, ?75(9): 2080-2085
52?Xiang L, Yu P, Zhang M, Hao J, Wang Y, Zhu L, Dai L, Mao L. Anal. Chem., ?2014, ?86(10): 5017-5023
53?JIANG Xiao?Jing, LIANG Rong?Ning, QIN Wei. Chinese J. Anal. Chem., ?2018, ?46(9): 1350-1356
姜曉晶, 梁榮寧, 秦 偉. ?分析化學, 2018, ?46(9): 1350-1356
54?He X, Zhang K, Li T, Jiang Y, Yu P, Mao L. J. Am. Chem. Soc., ?2017, ?139(4): 1396-1399
55?Zhang K, He X, Liu Y, Yu P, Fei J, Mao L. Anal. Chem., ?2017, ?89(12): 6794-6799
56?Colombo M L, McNeil S, Iwai N, Chang A, Shen M. J. Electrochem. Soc., ?2016, ?163(4): H3072-H3076
57?Colombo M L, Sweedler J V, Shen M. Anal. Chem., ?2015, ?87(10): 5095-5100
58?Hao J, Xiao T, Wu F, Yu P, Mao L. Anal. Chem., ?2016, ?88(22): 11238-11243
59?Wu F, Yu P, Mao L. Chem. Soc. Rev., ?2017, ?46(10): 2692-2704
60?Wu F, Cheng H, Wei H, Xiong T, Yu P, Mao L. Anal. Chem., ?2018, ?90(21): 13021-13029
61?CHENG Han, LAN Jia?Feng, WEI Guang?Han, HUANG Wei?Hua, CHENG Jie?Ke. Chinese J. Anal. Chem., ?2013, ?41(4): 540-545
程 寒, 蘭嘉峰, 韋光漢, 黃衛華, 程介克. ?分析化學, 2013, ?41(4): 540-545
62?HE Quan?Guo, LIANG Jing, LI Guang?Li, DENG Pei?Hong, LIU Jun, LIU Xiao?Peng. Chinese J. Anal. Chem., ?2018, ?46(3): 438-445
賀全國, 梁 靜, 李廣利, 鄧培紅, 劉 軍, 劉曉鵬. ?分析化學, 2018, ?46(3): 438-445
63?Liu X, Zhang M, Xiao T, Hao J, Li R, Mao L. Anal. Chem., ?2016, ?88(14): 7238-7244
64?Liu X, Xiao T, Wu F, Shen M, Zhang M, Yu H, Mao L. Angew. Chem.Int. Edit., ?2017, ?56(39): 11802-11806
65?YANG Li?Li, SONG Yi?Lin, XU Sheng?Wei, ZHANG Yu, XIAO Gui?Hua, ZHANG Song, GAO Fei, LI Zi?Yue, CAI Xin?Xia. Chinese J. Anal. Chem., ?2017, ?45(7): 1088-1095
楊麗麗, 宋軼琳, 徐聲偉, 張 禹, 肖桂花, 張 松, 高 飛, 李子岳, 蔡新霞. ?分析化學, 2017, ?45(7): 1088-1095
66?Kozai T D Y, Langhals N B, Patel P R, Deng X, Zhang H, Smith K L, Lahann J, Kotov N A, Kipke D R. Nat. Mater., ?2012, ?11(12): 1065-1073
67?Guan S, Wang J, Gu X, Zhao Y, Hou R, Fan H, Zou L, Gao L, Du M, Li C, Fang Y. Sci. Adv., ?2019, ?5(3): eaav2842
68?QIN Tai?Chun, LI Xiao?Gang, HAO Jie, YU Ping, MAO Lan?Qun. Chinese J. Anal. Chem., ?2015, ?43(3): 457-462
秦泰春, 李曉鋼, 郝 潔, 于 萍, 毛蘭群. ?分析化學, 2015, ?43(3): 457-462
69?LI Xiao?Gang, GUO Bin?Qian, QIN Tai?Chun, HAO Jie, YU Ping, MAO Lan?Qun. Chinese J. Anal. Chem., ?2016, ?44(9): 1465-1470
李曉鋼, 郭彬乾, 秦泰春, 郝 潔, 于 萍, 毛蘭群. ?分析化學, 2016, ?44(9): 1465-1470
70?SUN Jian?Hui, CAI Xin?Xia, LIU Jun?Tao, WANG Chun?Xing, LI Deng?Wang, CHEN Ze?Yuan, CHENG Chuan?Fu, WANG Jin?Hui, HU Dong?Mei. Chinese J. Anal. Chem., ?2017, ?45(4): 611-619
孫建輝, 蔡新霞, 劉軍濤, 王春興, 李登旺, 陳澤源, 程傳福, 汪金輝, 胡冬梅. ?分析化學, 2017, ?45(4): 611-619
Progress of in Vivo Electrochemical Analysis
of Brain Neurochemistry
XUE Yi?Fei1,2, XIAO Tong?Fang1, JIANG Ya?Nan1,2,
WU Fei1,2, YU Ping1,2, MAO Lan?Qun*1,2
1(Beijing National Laboratory for Molecular Sciences, Key Laboratory of Analytical Chemistry
for Living Biosystems, Institute of Chemistry, Chinese Academy of Sciences, Beijing 100190, China)
2(University of Chinese Academy of Sciences, Beijing 100049, China)
Abstract?Brain science has become one of the most advanced interdisciplinary research topics. Analysis of brain neurochemistry has attracted great attention in both neuroscience and chemistry, because it provides a powerful tool to understand the physiological and pathological progresses of the brain at a molecular level. For neurochemical monitoring, electrochemical methods come to prominence with high selectivity, sensitivity, spatiotemporal resolution and designable electrode/solution interface to match the pursuit for in vivo analysis of brain neurochemistry. This review summarizes the development of electrochemical approaches toward brain neurochemistry research from both principal and practical perspectives. In addition, future trends of in vivo electrochemical analysis are prospected.
Keywords?In vivo electrochemistry; Brain neurochemistry; In vivo measurement; Review
(Received 30 July 2019; accepted 16 August 2019)
This work was supported by the National Natural Science Foundation of China (Nos. 21790390, 21790391, 21621062, 21874139) and the National Key Research and Develop Program of China (No.2018YFE0200800).
