固定化胰蛋白酶整體小柱的制備及其用于微量蛋白質的快速酶解
2019-11-07 01:03:20鄭蒙蒙康經武
色譜 2019年12期
鄭蒙蒙, 韓 穎, 康經武,*
(1.中國科學院上海有機化學研究所, 生命有機化學國家重點實驗室, 上海 200032; 2.中國科學院大學, 北京 100049; 3.上海科技大學, 上海 201210)
近20年來,質譜法已成為大規模蛋白質分析的核心方法。基于質譜的蛋白組學分析通常采用自下而上(bottom-up)的策略[1],需要將完整蛋白質酶解成肽段后采用多維液相色譜-質譜分析得到肽段的氨基酸序列,通過搜庫檢索鑒定蛋白質[1,2]。在整個分析過程中,蛋白質樣品的高效酶解是蛋白質鑒定不可或缺的步驟。目前傳統的蛋白酶解方法為溶液酶解,整個過程需要持續12~24 h,耗時較長。因此,蛋白質的快速酶解方法受到研究者們的廣泛關注,其中包括如多酶系復合作用,培育篩選高效酶種,技術輔助酶解等,其中以固定化酶技術最具代表性[3,4]。與溶液酶解方法相比,固定化酶技術克服了游離酶穩定性低、難以與底物蛋白質分離等缺點。由于多孔材料的限閾作用[5],胰蛋白酶在微反應器中的局部濃度顯著提高,提升了酶解效率。
在眾多的固定化酶技術研究中,常用的載體有毛細管柱[6,7]、微流控芯片[8,9]、磁性微球[10-13]、納米多孔材料[14-16]和溶膠-凝膠[17]等。受材料性質的影響,酶固定化載體也存在自身的優缺點,如微流控芯片易于攜帶,小型化,可減少樣品與試劑的消耗量,但較難實現自動化[18];磁性微球雖然簡化了樣品分離過程,但微球本身易團聚,表面容易產生不可逆吸附,酶負載量低[10];納米多孔材料比表面積大,可固載大量的胰蛋白酶,但制備與化學修飾過程繁瑣[19]。
20世紀90年代初,Hjerten等[20]、Svec和Frechet[21]分別提出了“有機聚合物整體柱”的概念,隨后各類有機聚合整體柱被廣泛應用于固定化酶技術研究中[22-24]。與其他載體相比,整體柱材料具有較大的貫穿孔道和比表面積,其特有的對流性能可增加樣品的傳質效率,樣品與底物的接觸機會,以及酶催化的速率[25]。整體柱材料制備簡單,成本低廉,滲透性良好,機械強度高,是樣品處理和色譜分離的理想介質[26]。2002年,Svec課題組[22]以甲基丙烯酸羥乙酯(HEMA)、2-乙烯-4,4二甲基吖內酯(VAL)為功能單體,乙二醇二甲基丙烯酸酯(EDMA)為交聯劑在微流體通道內制備了poly(VAL-co-EDMA-co-HEMA)整體材料,用于胰蛋白酶的固定化及蛋白質的快速酶解。2013年,Dovichi課題組[27]將毛細管區帶電泳與ESI-MS串聯,并與固定化胰蛋白酶整體柱結合,成功地從3 ng小鼠單核巨噬細胞(RAW 264.7)的細胞裂解液中鑒定到7個蛋白質。同年,Krenkova等[28]將肽-N-糖苷酶F固定到修飾后的聚甲基丙烯酸酯整體柱上,用于多糖的分離檢測。但整體柱也存在分析通量不高的缺點,且催化酶大多通過與甲基丙烯酸縮水甘油酯(GMA)單體上的環氧基反應被固定在基質上[23,29],環氧基的開環衍生化處理使實驗變得耗時繁瑣。
為了解決以上問題,本研究發展了光聚合的方式批量制備固定化酶整體小柱用于蛋白質的快速酶解。該整體小柱采用4-戊烯酸琥珀酰亞胺酯(PAS)、HEMA為功能單體,可與交聯劑、致孔劑在20 μL的移液器吸頭尖端原位聚合形成整體柱床。酶分子的氨基與琥珀酰亞胺酯反應實現固定化,無需引入其他偶聯試劑與化學修飾。實驗采用離心輔助的方式對蛋白質樣品進行上樣、酶解、收集,簡化了樣品處理的流程,減少了樣品的損失與污染。整個酶解過程高效省時,可在10 min內完成,且離心式、平行化的樣品處理方式顯著地提高了樣品的分析通量,十分適用于微量蛋白質的快速酶解。
1 實驗部分
1.1 儀器、材料與試劑
Dionex Ultimate 3000高效液相色譜-LTQ-Fleet離子阱質譜儀(HPLC-MS/MS)、EASY-nLC 1200納升級液相色譜-Thermo Orbitrap Fusion Tribrid高分辨串聯質譜儀(Nano-LC-MS/MS)(美國Thermo Fisher Scientific公司); Mini spin高速離心機、Concentrator plus真空離心濃縮儀(德國Eppendorf公司); BT25S分析天平、PB-20標準型pH計(德國Sartorius公司); JY92-Ⅱ超聲波細胞破碎機(寧波新芝生物科技股份有限公司); Rotavapor R-210旋轉蒸發儀(瑞士Buchi公司); S-4800型掃描電子顯微鏡(日本日立株式會社)。
胰蛋白酶(trypsin)、二甲基亞砜(DMSO)、十二醇(1-dodecanol)、二硫蘇糖醇(DTT)、甲酸(HCOOH,色譜純)、碘代乙酰胺(IAM)(美國Sigma-Aldrich公司);蛋白酶抑制劑(德國Roche公司);乙腈和甲醇(色譜純,德國Merck公司);細胞色素C(CYC)、三羥甲基氨基甲烷(Tris)、尿素(上海阿拉丁生化科技股份有限公司);牛血清白蛋白(BSA)、碳酸氫銨、鹽酸(HCl)、N,N-二甲基甲酰胺(DMF)、二氯甲烷(CH2Cl2)、氨水(上海國藥集團化學試劑有限公司);季戊四醇三丙烯酸酯(PETA,美國Alfa Aesar公司); 1-乙基-(3-二甲基氨基丙基)碳酰二亞胺鹽酸鹽(EDC·HCl,上海安耐吉化學公司); 4-二甲氨基吡啶(DMAP,日本TCI公司);磷酸緩沖鹽溶液(PBS, pH=7.4)、Axygen?系列20 μL吸頭(上海生工生物工程股份有限公司); Sep-pack C18固相微萃取小柱(美國Waters公司); Pierce BCA蛋白定量試劑盒(美國Thermo Fisher Scientific公司)。Milli-Q去離子水由Millipore純水儀(美國Millipore公司)制備得到。
人急性早幼粒白血病(NB4)細胞與人急性T細胞白血病(Jurkat T)細胞由上海交通大學醫學院饋贈。
1.2 固定化胰蛋白酶整體小柱的制備
1.2.1整體小柱的制備
4-戊烯酸琥珀酰亞胺酯功能單體的制備:取4.5 gN-羥基琥珀酰亞胺,置于干燥的圓底燒瓶中,抽換氮氣3次后,加入150 mL二氯甲烷,攪拌均勻,加入3 mL戊烯酸,待白色顆粒溶解后再加入6.9 g EDC·HCl和4.0 g DMAP,室溫攪拌12 h,旋干,進行柱層析分離,旋轉蒸發濃縮后得到4-戊烯酸琥珀酰亞胺酯白色晶體5.57 g,產率為94.3%。
整體小柱的制備:取20 μL的移液器吸頭適量,分別于乙醇、丙酮中各超聲10 min,自然風干后,將柱頭封端。取0.090 5 g二苯甲酮[30],置于1 mL 1,4-丁二醇-甲醇(1∶1, v/v)中,混合均勻后用氮氣吹掃去氧。取10 μL上述溶液導入柱頭內,在氮氣保護下,于365 nm紫外燈下照射15 min,重復3次。將功能單體PAS和HEMA、交聯劑PETA、致孔劑DMSO、DMF和十二醇按質量分數5%、7.4%、12.6%、33%、8%和34%的比例混合均勻,取7 μL于柱頭中,在紫外燈裝置中光照30 min。聚合結束后切去柱頭的封端,用乙腈沖洗以除去未反應的功能單體和致孔劑,制得整體小柱,常溫保存。
1.2.2胰蛋白酶的固定化
用0.1 mol/L磷酸鹽緩沖鹽(pH 6.8)洗滌整體小柱3次,加入0.5 mg/mL 100 μL胰蛋白酶溶液(溶解于pH 6.8的磷酸鹽緩沖溶液),以2 000 r/min離心15 min,重復3次,然后用500 mmol/L Tris-HCl(pH 7.5)封閉未反應的PAS基團[19],于-20 ℃冰箱中保存待用。
1.3 樣品制備與處理
1.3.1NB4細胞與Jurkat T細胞培養
NB4細胞與Jurkat T細胞均接種于含體積分數10%的胎牛血清、100 U/mL青霉素、100 U/mL鏈霉素的RPMI-1640培養液中,置于37 ℃、含5%(體積分數) CO2的飽和濕度孵箱中懸浮培養。每隔48 h更換新的培養基,傳代培養。
1.3.2固定化酶整體小柱酶解標準蛋白質與細胞樣品
采用固定化酶整體小柱酶解標準蛋白質時,整體小柱先用50 mmol/L碳酸氫銨溶液洗滌3次,除去未鍵合的胰蛋白酶。取適量CYC與BSA,置于50 mmol/L碳酸氫銨緩沖液中,分別配制成200 ng/μL的CYC標準溶液和BSA標準溶液,各取100 μL于固定化酶整體小柱內,以2 000 r/min離心酶解10 min,收集酶解液,進行HPLC-MS/MS分析。
在酶解細胞樣品時,配制10 mL裂解緩沖液(4.804 8 g尿素和0.039 5 g碳酸氫銨,以及一片蛋白酶抑制劑片劑溶于10 mL Milli-Q H2O中)。分別收集1×105個NB4細胞與Jurkat T細胞,用預冷的PBS緩沖液清洗3次,加入100 μL預冷的裂解緩沖液,在冰上以60 W功率超聲破碎2 min。然后以9 780 r/min離心10 min,收集上清液。采用Pierce BCA蛋白定量試劑盒對細胞裂解液進行定量。向蛋白質溶液中加入終濃度為10 mmol/L的DTT溶液,置于37 ℃溫育1 h,隨后加入終濃度為5 mmol/L的IAM,并置于暗室中常溫反應45 min。向蛋白質溶液中加入50 mmol/L碳酸氫銨,將尿素濃度稀釋至1 mol/L。取稀釋后的細胞裂解液于固定化酶整體小柱中,以2 000 r/min離心酶解10 min,收集酶解液,通過Sep-pack C18固相微萃取柱脫鹽。酶解所得肽段進行Nano-LC-MS/MS分析。
1.3.3溶液酶解標準蛋白質與細胞樣品
取適量CYC和BSA于50 mmol/L碳酸氫銨緩沖液中,分別配制成200 ng/μL的CYC標準溶液和BSA標準溶液,于100 ℃變性處理10 min。待標準樣品冷卻至室溫,以蛋白質與酶質量比為40∶1的比例加入胰蛋白酶,置于37 ℃水浴中酶解16 h。酶解結束后置于-20 ℃冰箱內保存備用。酶解所得肽段進行HPLC-MS/MS分析。
在酶解細胞樣品時,預處理步驟同1.3.2節,1×105個NB4細胞與Jurkat T細胞經超聲破碎、還原烷基化、樣品稀釋后,在樣品溶液中以蛋白質與酶質量比為40∶1的比例加入胰蛋白酶,置于37 ℃水浴中酶解16 h。酶解結束后通過Sep-pack C18脫鹽。酶解所得肽段進行Nano-LC-MS/MS分析。
1.4 分析條件
1.4.1標準蛋白質樣品
標準蛋白樣品酶解后由Dionex Ultimate 3000液相色譜與LTQ-Fleet離子阱質譜聯用系統進行肽段的分離與鑒定。
色譜條件:分析色譜柱為Agilent Zorbax SB-C18(250 mm×4.6 mm, 5 μm);柱溫為25 ℃;流動相A相為0.1%(v/v)甲酸水溶液,B相為0.1%(v/v)甲酸乙腈溶液;流速為0.5 mL/min;梯度洗脫程序:0~25 min, 5%B~45%B; 25~30 min, 45%B~72%B; 30~40 min, 72%B。進樣量為10 μL(標準蛋白質的質量濃度為200 ng/μL)。
質譜條件:離子源為電噴霧電離源,正離子模式;離子源電壓為3.0 kV;金屬毛細管溫度為275 ℃;一級質譜掃描范圍為m/z300~1 800;二級質譜中母離子的選擇采用數據依賴模式;碰撞誘導解離(CID)能量為35 eV。
數據檢索條件:由MaxQuant(1.5.4.1)軟件進行檢索,蛋白酶切類型選擇胰蛋白酶,允許最大肽段漏切位點數為2。因BSA采用了高溫加熱的方式進行蛋白質變性預處理,檢索時無固定修飾,蛋氨酸上的氧化(Oxidation, M)與蛋白質N端的乙酰化(Acetyl, proteinN-term)設置為可變修飾。允許的最小肽長度設置為4。肽段水平假陽性率(FDR)≤1%。
1.4.2細胞樣品
細胞樣品酶解后由EASY-nLC 1200與Thermo Orbitrap Fusion Tribrid質譜聯用系統進行分離與鑒定。
色譜條件:色譜柱為實驗室自主裝填的Nano-C18柱(15 cm×75 μm, 3 μm,填料來自德國Dr.Maisch公司);柱溫為25 ℃;流動相A相為0.1%(v/v)甲酸水溶液,B相為80%(v/v)乙腈水溶液(含0.1%(v/v)甲酸);流速為300 nL/min;梯度洗脫程序:0~50 min, 0%B~30%B; 50~53 min, 30%B~38%B; 53~54 min, 38%B~90%B; 54~60 min, 90%B。進樣量:1 μL(肽段質量約為200 ng)。
質譜條件:離子源為電噴霧電離源,正離子模式;離子源電壓為2.3 kV;一級質譜分辨率為120 000,一級質譜的自動增益控制(AGC)為5×105,最大離子注入時間為50 ms,全掃描范圍為m/z300~1 400;二級質譜中母離子的選擇采用數據依賴模式;高能誘導裂解(HCD)的歸一化碰撞能量為28%,二級AGC為5×104,動態質量排除窗口為30 s。
數據檢索條件:由MaxQuant(1.5.4.1)軟件進行檢索,蛋白酶切類型選擇胰蛋白酶,允許最大肽段漏切位點數為2。半胱氨酸上的氨基甲酰甲基化(carbamidomethyl)為固定修飾,蛋氨酸上的氧化與蛋白質N端的乙酰化為可變修飾。允許的最小肽長度設置為7。肽段水平FDR≤1%。鑒定到的細胞蛋白數量為單次實驗的結果。
2 結果與討論
2.1 固定化酶整體小柱的制備及表征
在制備有機聚合整體柱時,因GMA在聚合后仍含有較活潑的環氧基而頗受青睞[23,29],但環氧基的化學改性需要在強堿性或高溫下進行,條件比較嚴苛,于是本文選用了更活潑的PAS作為功能單體之一,其在常溫下即可與氨基等活性基團反應,易于配體修飾。為了降低水解肽段的非特異性吸附[15],實驗選用親水性的PETA作為交聯劑,同時在聚合溶液中加入功能單體HEMA增加整體材料的親水性。三元致孔劑DMSO、DMF、十二醇的使用可以得到良好的整體骨架與孔結構。
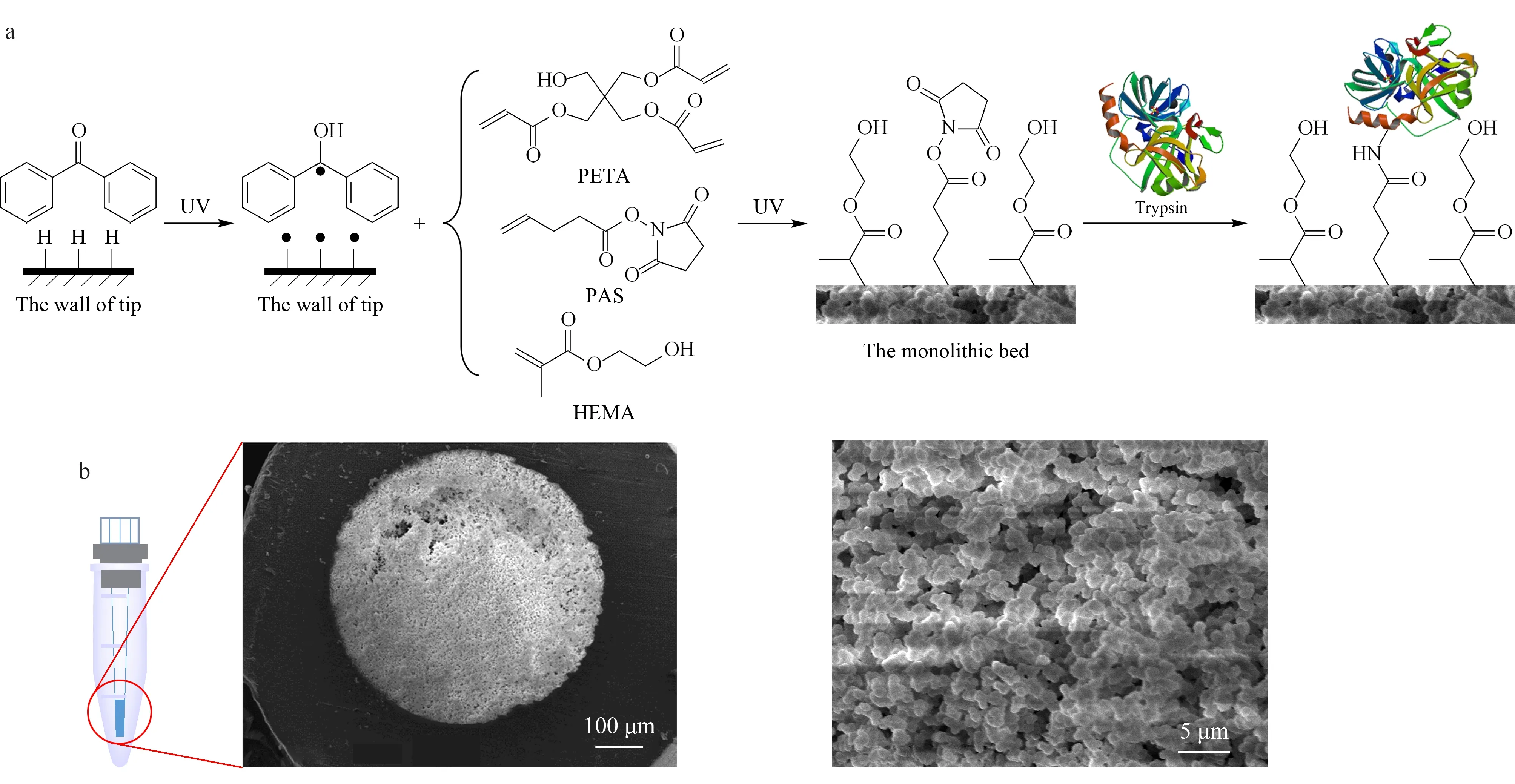
圖1 固定化胰蛋白酶整體小柱的(a)制備流程圖和(b)掃描電鏡圖
本研究采用光聚合的方式制備整體小柱,聚合過程如圖1a所示:在紫外光照射下,光敏劑二苯甲酮從移液器吸頭的聚丙烯表面奪氫生成半頻哪醇自由基,并與吸頭內表面的自由基結合形成穩定的表面引發劑。在柱頭加入聚合液后,再次進行紫外燈照射,表面引發劑被激活,釋放出自由基,實現功能單體與小柱內表面的交聯接枝反應。整個光聚合反應僅需2 h便可完成。整體小柱切面的掃描電子顯微鏡(SEM)圖見圖1b。可以看出,柱體材料與移液器吸頭內表面結合緊密,無脫壁、塌陷等現象,通過觀察SEM放大圖,該材料具有較大的貫穿孔道,既保證了胰蛋白酶的穩定固定化,又保證了良好的滲透性,使溶液在柱床中進行對流傳質。
2.2 胰蛋白酶固載量的測定
胰酶的固載量是影響酶解效率的關鍵因素[5]。在特定的限閾范圍內,胰酶的固載量越大,其局部濃度越大;與底物蛋白分子接觸的機會越大,則酶解效率越高[31]。實驗用Pierce BCA試劑盒測定固定化前后緩沖溶液中胰蛋白酶的質量,以確定整體小柱上胰酶的固載量。
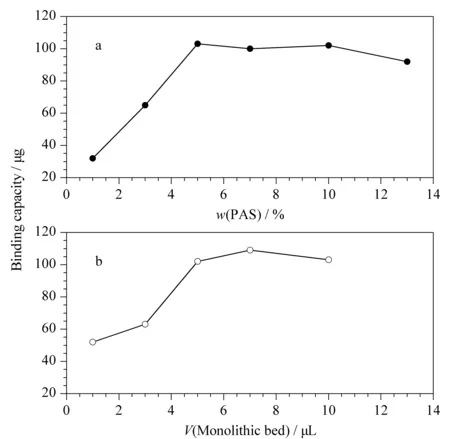
圖2 不同(a)PAS質量分數和(b)柱床體積對胰蛋白酶固載量的影響
實驗首先考察了PAS的質量分數對胰酶固載量的影響,在保持功能單體(12.4%)、交聯劑(12.6%)與致孔劑(75%)所占質量分數不變的情況下,調節功能單體 PAS與 HEMA的比例,制備了6款整體小柱。如圖2a所示,當聚合溶液中活性酯PAS的質量分數從1%增至5%時,酶的固載量逐漸上升;活性酯的質量分數從5%繼續增加至13%時,酶的固載量略有降低。親水性的聚合材料可以降低水解肽段的非特異性吸附,在聚合溶液中加入HEMA可增加整體材料的親水性。實驗最終確定功能單體PAS與HEMA的質量分數分別為5%和7.4%,在確保胰酶最大固載量的同時減少了水解肽段的非特異性吸附。
隨后實驗考察了整體小柱的柱床體積對酶固載量的影響,結果見圖2b。隨著柱床體積增加,胰酶的固載量也逐漸增加。但當柱床體積達到10 μL時,整體小柱在酶固定化過程中出現了明顯的脫壁現象。實驗最終選用7 μL的柱床體積,此時胰酶的固定量為108.8 μg。
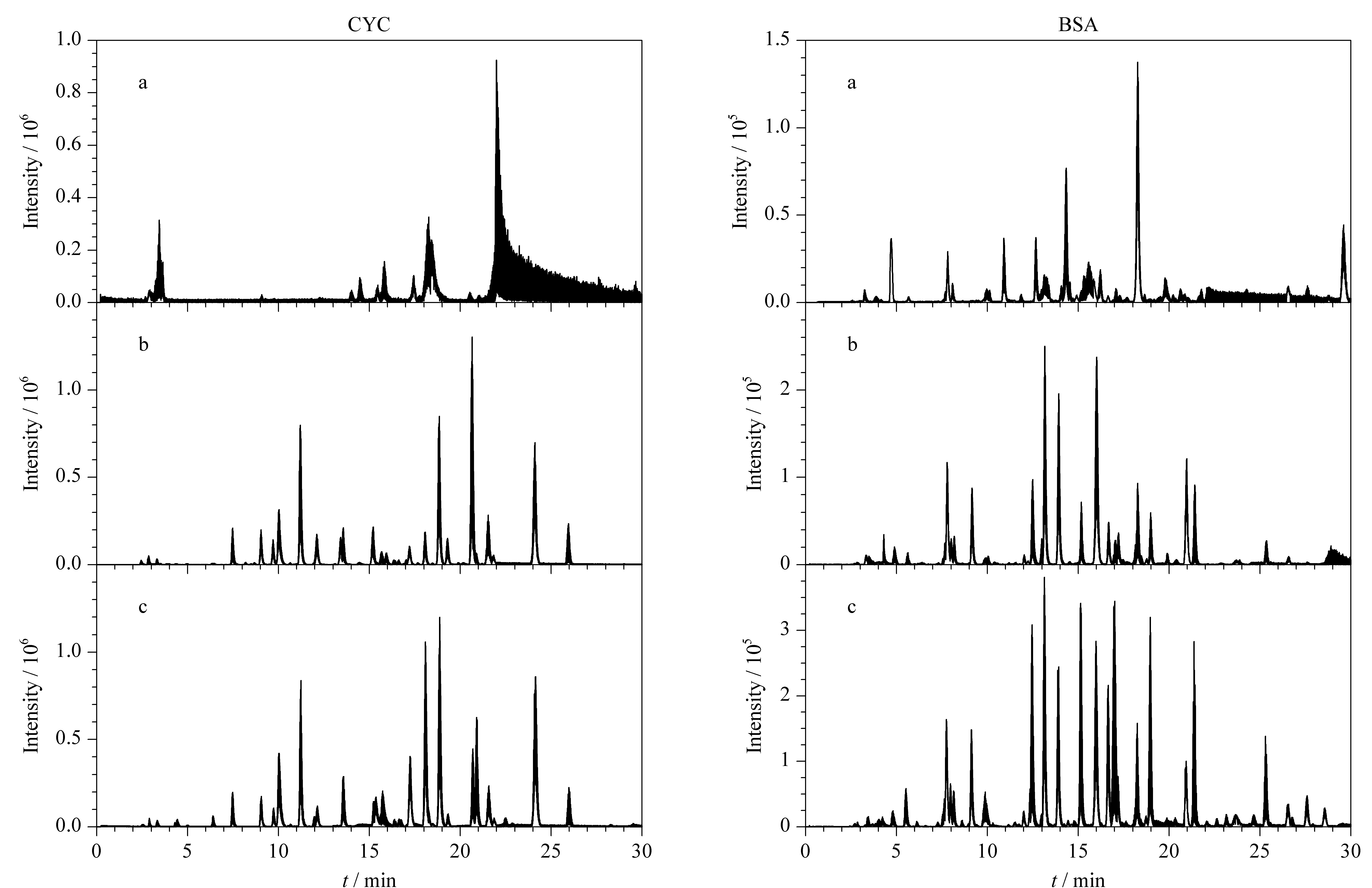
圖3 CYC和BSA經(a)溶液酶解10 min、(b)固定化胰蛋白酶整體小柱酶解10 min、(c)溶液酶解16 h后所得肽段的色譜圖
2.3 固定化酶整體小柱用于標準蛋白質的高效酶解
在均相酶解過程中,傳質速率是限制酶解動力學過程的主要因素之一[31]。大多數常用的內肽酶和外肽酶的米氏常數(Km)值都在5~50 mmol/L之間,當蛋白質濃度低于μmol/L水平時,酶解過程將受到顯著的阻礙[32]。為了避免這類濃度極低卻又重要的痕量蛋白質丟失,可以設法提高胰酶與蛋白質的相對濃度。在固定化酶整體小柱中,酶分子以共價鍵合的方式固定在柱體的貫通孔道內,相對于游離酶而言,其局部濃度得到了很大提升,當樣品上樣后,待酶解蛋白質與胰蛋白酶的相對濃度也進一步增加[31]。同時離心帶來的驅動力可以將所有流動相強制通過多孔介質,達到最大的對流效果[33],酶解效果也隨之增強。
實驗首先考察了不同酶解時間對固定化酶整體小柱酶解效率的影響。當酶解時間為5 min時,200 ng/μL的CYC標準溶液酶解并不完全;而將酶解時間增加至10 min時,酶解效果良好;當酶解時間為15 min時,酶解效率與10 min條件下的效果相當。因此選用10 min作為優化后的酶解時間。
在優化酶解時間后,分別取100 μL 200 ng/μL的CYC和BSA標準溶液于固定化酶整體小柱中酶解10 min。同時,取相同濃度與體積的CYC和BSA標準溶液于37 ℃恒溫條件下分別溶液酶解10 min與16 h。不同酶解條件下得到的酶解液分別進行HPLC-MS/MS分析,所得色譜圖見圖3。對比圖3a與圖3b可以發現,當酶解時間為10 min時,固定化酶整體小柱的酶解效果明顯優于溶液酶解。CYC標準溶液通過固定化酶整體小柱酶解可鑒定出17條肽段,對應的氨基酸序列覆蓋率為85%,溶液酶解10 min僅能鑒定到2條肽段,且在圖3a的22 min處可明顯觀察到CYC蛋白的信號峰。當溶液酶解時間增至16 h時(見圖3c),酶解效率提升,可以鑒定到17條肽段。
將BSA標準溶液恒溫溶液酶解10 min(見圖3a),僅可鑒定到4條肽段,序列覆蓋率僅為5.8%,但將BSA通過固定化酶酶解10 min(見圖3b)后可鑒定到37條肽段,氨基酸序列覆蓋度為43%,與16 h的溶液酶解(見圖3c)效果基本一致。但溶液酶解由于酶解時間過長,容易發生肽段非特異性裂解,胰酶自酶解等現象[2],在色譜圖中產生信號干擾峰。而整體小柱的孔道可以穩定胰蛋白酶,提供較高的局部酶濃度,在縮短酶解時間的同時有效避免了自酶解干擾。
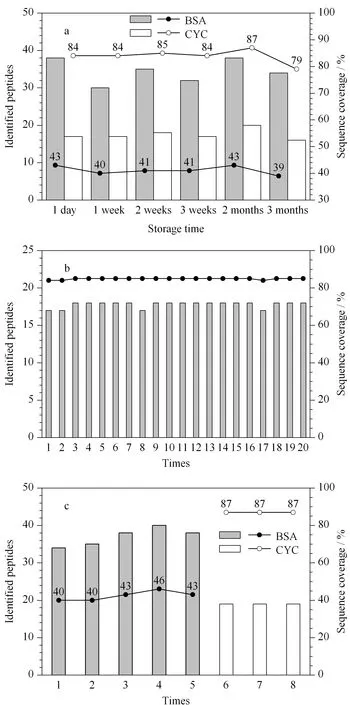
圖4 不同條件下固定化胰蛋白酶整體小柱酶解CYC和BSA得到的肽段數量和序列覆蓋率
由于細胞中的蛋白質呈現高動態分布,許多蛋白質在低豐度下自然表達,這類低濃度微量蛋白的酶解是蛋白組學研究中遇到的不利因素[34]。為了考察固定化酶整體小柱對微量蛋白質的酶解效果,實驗選擇了質量濃度為1 ng/μL與10 ng/μL的CYC標準溶液作為樣品,經固定化酶整體小柱酶解后進行HPLC-MS/MS分析。實驗結果表明,當CYC含量低至1 ng/μL時,仍能鑒定到5條肽段,對應的氨基酸序列覆蓋度為24%;當含量提高至10 ng/μL時,成功鑒定到12條肽段,對應的序列覆蓋度為62%。說明固定化酶整體小柱對低濃度標準蛋白仍具有較好的酶解效果。
2.4 固定化酶整體小柱穩定性和重復性考察
本文還考察了固定化酶整體小柱的穩定性。將固定化酶整體小柱于-20 ℃儲存1 d、1周、2周、3周、2個月和3個月后取出,恢復至室溫后用于CYC和BSA標準溶液的酶解(200 ng/μL)。實驗結果如圖4a所示,即使儲存了3個月,該整體小柱仍維持著較高的酶解性能,在酶解產物中分別鑒定到了16條CYC肽段和34條BSA肽段,酶解效率較新鮮制備的整體小柱僅有輕微的降低,說明固定化可以穩定胰蛋白酶的性能。
為了考察固定化酶整體小柱的可重復使用性,實驗選取了同一支固定化酶整體小柱先后酶解了20個200 ng/μL的CYC標準溶液,其中,每兩個相鄰樣品間均用50 mmol/L碳酸氫銨溶液清洗2次。得到的實驗結果如圖4b所示:同一支整體小柱即使重復使用了20次,酶解效率依然沒有降低,第20次實驗中仍能鑒定到17條肽段,對應氨基酸序列覆蓋率高達85%。考慮到同一支固定化酶整體小柱重復處理不同樣品時可能會存在樣品殘留引起的記憶效應。本實驗取同一支整體小柱依次酶解5批200 ng/μL的BSA標準溶液,隨后采用這支整體小柱酶解3批200 ng/μL的CYC標準溶液(見圖4c)。在分析BSA時,整體小柱維持了穩定的酶解性能,5次酶解鑒定到的肽段數量相當,氨基酸序列覆蓋度為40%~46%;分析CYC時,搜庫結果顯示沒有屬于BSA的肽段,重復使用的整體小柱對CYC仍有較高的酶解效率,對應蛋白質的氨基酸序列覆蓋率高達87%。以上結果說明,固定化酶整體小柱對肽段的非特異性吸附較低,可重復用于酶解不同的蛋白質樣品。
2.5 固定化酶整體小柱酶解NB4與Jurkat T細胞
為了考察固定化酶整體小柱對復雜樣品的酶解效率,本文選用了NB4細胞與Jurkat T細胞進行實驗。分別收集1×105個NB4與Jurkat T細胞,超聲破碎、還原烷基化后分別導入整體小柱內,以2 000 r/min離心10 min后收集酶解液,經脫鹽處理后進行Nano-LC-MS/MS分析。作為對照,相同數量的細胞樣品經相同條件處理后于37 ℃溶液酶解16 h,收集的酶解液經脫鹽后由同一儀器相同參數下分析。
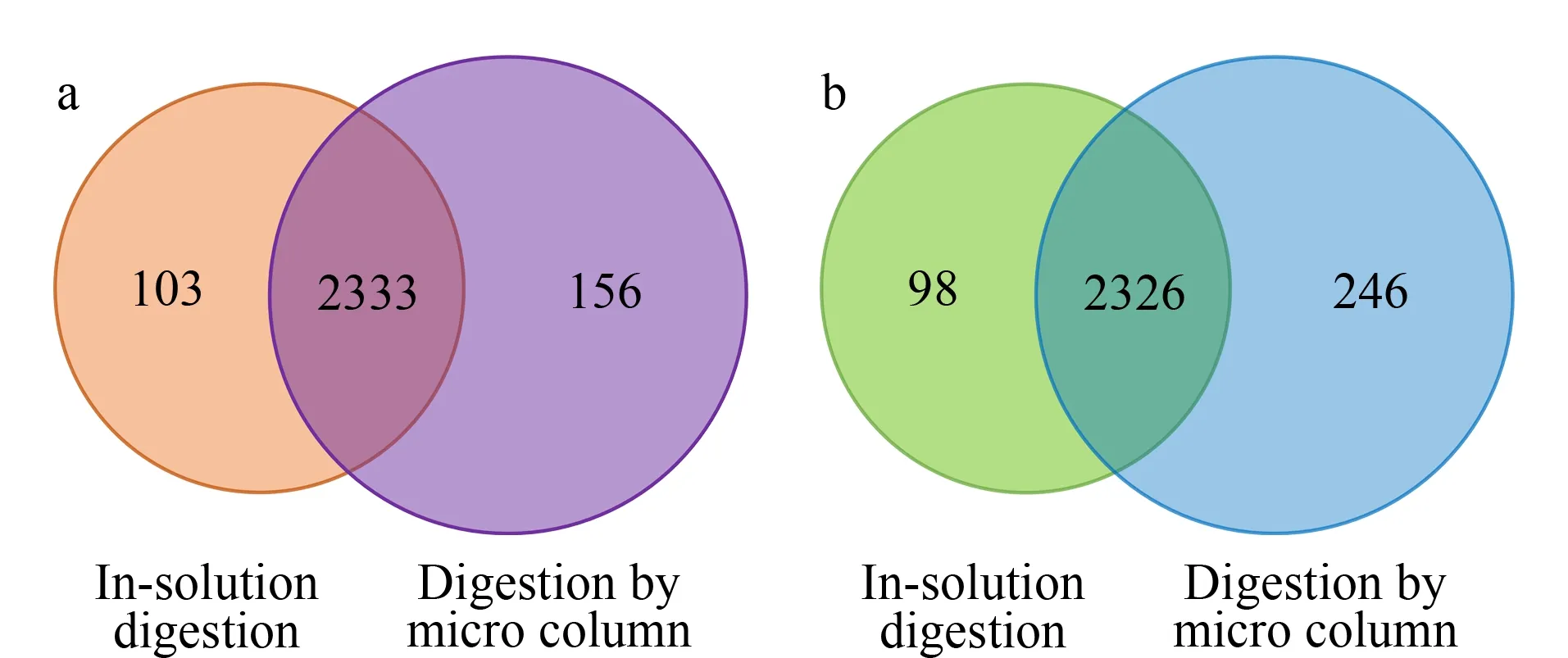
圖5 兩種方法酶解(a)NB4細胞和(b)Jurkat T細胞所得蛋白質數量的文氏圖
圖5a顯示了NB4細胞蛋白質組分析結果,從文氏圖可以看出,兩種酶解方法共同鑒定到2 333個蛋白質,固定化酶整體小柱鑒定到的蛋白質總數為2 489個,大于溶液酶解得到的蛋白質數量(2 436個),提高了2.2%。對于Jurkat T細胞而言,整體小柱鑒定的蛋白質總量為2 572個,較溶液酶解鑒定到的蛋白質數量提高了6.1%。在胰蛋白酶的固定化過程中,偶爾會發生酶解活性位點被堵塞、胰酶三級結構發生扭曲等現象[35],而對于這兩種細胞系而言,溶液酶解與固定化酶酶解兩種方式鑒定到的蛋白質重疊部分占比較大,說明了胰蛋白酶在固定過程中并未過多改變自身的構象與活性,固定化后仍然維持著穩定的酶解性能和高效的酶解效率。
以上分析結果體現了固定化酶整體小柱在復雜蛋白質樣品中應用的可行性。復雜生物樣品在整體小柱中的酶解時間也僅需10 min,鑒定到的蛋白質數量優于溶液酶解方法。且離心式、平行化的酶解過程簡化了樣品處理的流程,減少了樣品的損失與污染,同時增加了樣品處理的通量,十分適用于高通量蛋白組學及臨床醫學的研究。
3 結論
在基于質譜的蛋白質組學分析中,樣品制備是蛋白質分析的關鍵步驟,而蛋白質的酶解效率將影響整個樣品前處理的進程。本研究以光聚合法在20 μL移液器吸頭原位聚合形成整體小柱用于微量蛋白質的快速酶解。該固定化酶整體小柱使用4-戊烯酸琥珀酰亞胺酯為功能單體,無需化學修飾與其他偶聯試劑即可一步實現胰蛋白酶的固定化。同理,該固定化技術有望應用于其他不同特性的酶。制備得到的整體小柱機械強度高,化學性能穩定。在酶解過程中,樣品消耗量低,分析通量高,十分適用于微量的蛋白組學樣品分析。將固定化酶整體小柱應用于酶解NB4細胞與Jurkat T細胞,酶解效果優于常規的溶液酶解,在蛋白質組學分析領域展現出較大的應用潛力。