基于色譜法與質譜法分析唾液酸的衍生方法的研究進展
2019-11-07 01:03:18張啟偉
色譜 2019年12期
關鍵詞:方法
張啟偉, 鄭 琦
(江漢大學化學與環境工程學院, 交叉學科研究院, 光電化學材料與器件教育部重點實驗室, 湖北 武漢 430056)
唾液酸(sialic acids)是神經氨酸(neuraminic acid)的N-或O-取代衍生物,擁有相同的9碳核心結構,在自然界中已發現60余種[1,2]。神經氨酸的N-或O-取代主要發生在C4、C5、C7、C8、C9位置,其中依據C5位置取代基團的不同,唾液酸被分成3大類,即N-乙酰神經氨酸(N-acetylneuraminic acid, NeuAc)、N-羥乙酰神經氨酸(N-glycolylneuraminic acid, NeuGc)和酮基-脫氧壬酮糖酸(2-keto-3-deoxynononic acid, KDN);發生在其他位置的取代基團主要包括乙酰基、磺酸基、乳酰基、甲基和磷酸基等[3,4]。3種結構最簡單的唾液酸如圖1所示,其他唾液酸均為此3種唾液酸的衍生物。
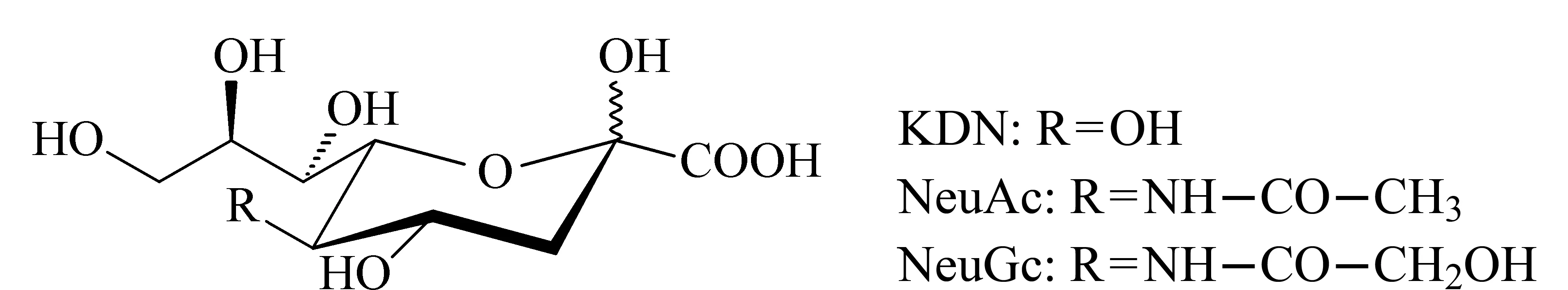
圖1 唾液酸的基本結構
唾液酸大量存在于包括人體和各種脊椎動物體內,也存在于部分無脊椎動物、真菌和細菌中[3,5-9]。唾液酸有著廣泛的生物功能,可參與到細胞間相互作用、細胞黏附與分化、神經發育與記憶形成、腸道微生態平衡等生理過程;與此同時,也與炎癥、癌癥、傳染病、神經退行性疾病、自身免疫疾病等病理性活動密切相關[10-14]。為了深入理解唾液酸的生物功能,糖組學領域的科研工作者經過幾十年的努力,陸續建立了多種表征分析方法。
以色譜法與質譜法為核心的檢測技術,是當前唾液酸表征分析研究中最主要且應用最廣泛的方法[15-17]。然而在通過色譜法、質譜法分析天然唾液酸時,經常遭遇檢測靈敏度低、結構易被破壞、色譜分離度差等問題,因此在樣品前處理過程中,通常需要將唾液酸衍生化。衍生化的優勢包括提高檢測靈敏度,穩定唾液酸結構,增強色譜分離度,改變碎片化模式,強化結構特征等。本文將重點對用于色譜與質譜分析唾液酸的衍生方法進行歸納和總結,并展望該領域的應用前景及發展趨勢等。
生物體內的唾液酸,一部分以游離的形式存在;另一部分則經常作為糖綴合物(glycoconjugates)的組成結構,以多種方式連接于其末端[18,19]。這使得關于唾液酸的色譜與質譜分析方法大體上可分成4個層次,即單糖水平、寡糖水平、糖肽水平和糖蛋白水平。目前而言,在糖肽及糖蛋白等高水平上表征唾液酸仍然面臨極大困難,因而大部分衍生方法主要應用于單糖與寡糖水平上。本文將從單糖、游離唾液酸、N/O-聚糖、糖脂等4個層面總結唾液酸的衍生方法。
1 單糖水平的唾液酸衍生方法
1.1 鄰苯二胺類試劑
連接于糖綴合物上的唾液酸能夠通過酸解、酶解等方法被解離[20,21]。解離后,唾液酸分子中的α-酮基-羧基能夠與鄰苯二胺類試劑的氨基發生反應,從而形成一種喹喔啉衍生物(見圖2)。在水溶液中,唾液酸能夠以α-吡喃糖、β-吡喃糖、非環狀酮等多種形式存在(以β-吡喃糖形式為主)[22]。而上述衍生方法的一個重要優勢在于反應生成的喹喔啉衍生物能夠消除端基異構體,防止單一結構在色譜分離時因異頭碳而產生多色譜峰。因衍生物具有熒光效應,故此方法常用于唾液酸的液相色譜-熒光檢測分析中[15]。與此同時,衍生后的唾液酸能夠呈現出更好的色譜分離度和質譜電離效率,使得其在唾液酸的液相色譜-質譜分析中也有較為廣泛的應用[3,15,23]。

圖2 唾液酸與鄰苯二胺反應生成喹喔啉衍生物的示意圖
已報道的鄰苯二胺類試劑主要包括鄰苯二胺(o-phenylenediamine, OPD)、3,4-二氨基甲苯(3,4-diaminotoluene, DAT)、1,2-二氨基-4,5-二甲氧基苯(1,2-diamino-4,5-dimethoxybenzene, DDB)、4,5-二甲基-1,2-苯二胺(4,5-dimethylbenzene-1,2-diamine, DMBA)、4,5-亞甲二氧基-1,2-苯二胺(4,5-methylenedioxy-1,2-phenylenediamine, DMB)等(見圖2)[24-28]。其中,DMB因具有熒光信號強、能夠顯著提高色譜分離度等優勢,成為在單糖水平上衍生唾液酸的首選衍生試劑[15],相關方法已經被開發成商業化試劑盒。相較于DMB,其他鄰苯二胺類試劑具有價格低廉、易獲得同位素標記物等優勢,同樣具有重要的應用價值。值得注意的是,此類試劑也可以和其他α-酮酸發生反應,例如α-酮戊二酸、丙酮酸、對羥基苯丙酮酸等。因此在對生物樣品進行直接衍生分析時需要注意信號干擾等問題[15]。
鄰苯二胺類試劑衍生的唾液酸在串聯質譜分析中能夠產生特征性碎片離子。以被DMB衍生的唾液酸為例,其可以產生m/z313、295、283、229等碎片離子,有利于唾液酸的鑒別[3,29]。此類試劑衍生的唾液酸,即便含有不同的修飾基團,其碎片化模式依然非常相似,難以通過質譜有效辨別不同結構。因而在鑒定同分異構體、修飾基團的連接位置等方面,此方法應用性不強。
鄰苯二胺類試劑衍生方法的主要缺陷有兩點:樣品濃度會隨著反應時間的延長而呈現下降趨勢,這意味著衍生過程伴隨著未知的副反應;同一條件對不同唾液酸的衍生效率并不完全相同,存在衍生歧視的風險[28,30-32]。這些問題導致此類試劑在唾液酸混合物的定量分析研究中存在局限性。

圖3 苯胺類試劑衍生唾液酸的示意圖
1.2 苯胺類試劑
標記唾液酸的代表性苯胺類試劑為2-氨基吖啶酮(2-aminoacridone, AMAC),其通過還原胺化作用與唾液酸的α-酮基發生反應[33,34]。然而,常規的還原胺化反應條件易引起脫羧作用,從而導致生成大量相對分子質量減少44的產物,即衍生后的樣品中既包含完整產物,也存在中性丟失產物(見圖3),嚴重制約了此方法的應用。為了盡可能提高產物的單一性,Szabo等[34]通過改變反應條件以提高脫羧作用發生的概率,從而只生成中性丟失產物。除AMAC外,7-氨基萘-1,3-二磺酸(7-aminonaphtalene-1,3-disulfonic acid)、4-氨基苯磺酸(4-aminobenzene sulfonic acid)等苯胺類試劑也已被用于衍生唾液酸。不同之處在于,這些試劑的磺酸基具有強負電荷屬性,主要用于毛細管電泳以及負離子模式下的質譜分析[35,36]。
1.3 甲基化試劑
在強堿性條件下,通過碘甲烷(methyl iodide)將分子中羥基、羧基、氨基上的氫原子用甲基取代,是最早建立的唾液酸衍生方法之一[37],一般稱之為全甲基化方法。全甲基化方法自建立后又多次被優化,目前仍然是衍生寡糖的重要方法之一[16]。另一類甲基化方法則先衍生羧基,然后再衍生羥基等其他基團。例如,在用氯化氫的甲醇溶液將羧基甲基化后,可以通過乙酸酐吡啶溶液(acetic anhydride pyridine)、雙(三甲基硅烷)三氟乙酰胺(bis(trimethylsilyl)trifluoroacetamide)等試劑將唾液酸分子乙酰化或硅烷化[38,39]。但是自鄰苯二胺類試劑被廣泛應用后,上述方法已較少用于單糖水平上的唾液酸分析,而且苛刻的反應條件易破壞唾液酸的某些修飾基團[40],進一步限制了相關方法的應用。
另有一種兩步衍生方法,先用重氮甲烷(diazomethane)衍生羧基后,再用七氟丁酸(heptafluorobutyric acid)衍生羥基。該反應的生成物既可以保留唾液酸的修飾基團(如乙酰基、甲基等),又可在氣相色譜-質譜分析中產生特征性碎片離子,有利于鑒定修飾基團的連接位置以及同分異構體等[41,42]。此方法具有反應速率高且生成物穩定性強等優勢,但由于重氮甲烷存在不穩定性和高危險性,使其應用受到了限制。
1.4 烷基胺類試劑
苯胺類試劑主要和唾液酸的α-酮基發生反應,而烷基胺類試劑則通常被用于衍生羧基,已報道的試劑包括十七氟癸胺(heptadecafluoroundecylamine, HFUA)、4-(N,N-二甲基氨基)-7-(2-氨基乙基氨基)-2,1,3-苯并惡二唑(4-(N,N-dimethylaminosulfonyl)-7-(2-aminoethylamino)-2,1,3-benzoxadiazole)、4-(N,N-二甲基氨基)-7-哌嗪子基-2,1,3-苯并惡二唑(4-(N,N-dimethylaminosulfonyl)-7-piperazino-2,1,3-benzoxadiazole)等[43-45]。此類試劑能夠同時衍生唾液酸及其氧化產物,例如NeuAc及其氧化產物4-(乙酰氨基)-2,4-二脫氧-D-甘油-D-半乳糖辛酸(4-(acetylamino)-2,4-dideoxy-D-glycero-D-galacto-octonic acid, ADOA)可以用于評估生物體遭受的氧化壓力等[44]。以HFUA為代表的含氟衍生試劑,因其衍生物可以特異性地保留在含氟固定相的色譜柱上,并與樣品中的其他非氟組分分離,故而能夠有效減少液相色譜以及LC-MS分析中的干擾信號[45]。
與苯胺類試劑相似,烷基胺類試劑無法消除端基異構體,使得單一唾液酸易形成多個色譜峰,大大增加了色譜圖的復雜性[45]。先消除端基異構體再衍生唾液酸是一種可行的方案。已有文獻[46]報道,唾液酸能夠被雙氧水氧化從而轉變為相對分子質量減少28的產物,即ADOA,此產物無端基異構體,或許可以在胺類試劑衍生唾液酸的研究中發揮重要作用。
2 游離唾液酸的衍生方法
糖綴合物上的唾液酸在衍生反應過程中可能發生解離,從而干擾游離唾液酸的定量分析,而使用苯二胺類試劑衍生游離唾液酸的關鍵在于選擇合適的反應條件以抑制解離的發生。最常用的鄰苯二胺類試劑是DMB,典型的衍生條件是在酸性環境下保持50 ℃恒溫2.5 h;改變條件至4 ℃并延長反應時間至48 h,即可有效衍生樣品中游離的唾液酸[18]。DMBA也可以通過類似方法抑制解離并衍生游離的唾液酸[47]。
上述方法現已成為分析游離唾液酸的常規策略,除此之外,近些年也有其他方法被報道[27,48]。氯化氫的正丁醇溶液可以直接衍生游離唾液酸,此反應在無水條件下進行以抑制糖綴合物上的唾液酸發生解離[48]。先從樣品中分離游離的唾液酸,再進行衍生化處理,這也是一種有效的分析策略[27,49]。雖然此方法操作復雜,但適用性更廣泛。采取簡化前處理的方式,通過在負離子模式下的質譜分析也可以直接分析游離的唾液酸[50]。
3 在N/O-聚糖水平上的唾液酸衍生方法
寡糖是一種由幾個至幾十個單糖單元聚合而成的碳水化合物,經常作為糖綴合物的重要組成部分通過多種途徑參與生物體的生理功能。一般而言,可以將寡糖分為4大類:N-聚糖(N-glycan)、O-聚糖(O-glycan)、糖胺聚糖(glycosaminoglycan)和糖脂(glycosphingolipid)[51]。其中,糖胺聚糖很少被唾液酸化(sialylation),目前僅在硫酸角質素(keratan sulfate)中發現了此現象[52-54];其他寡糖則經常發生唾液酸化。因此,本文不討論糖胺聚糖,僅從N-聚糖、O-聚糖和糖脂的層面總結在寡糖水平上的唾液酸衍生方法。
N-聚糖連接于蛋白質的天冬酰胺殘基上,O-聚糖主要連接于蛋白質的絲氨酸或蘇氨酸殘基上,二者結構較為相似,衍生方法基本可以通用。在N/O-聚糖水平上的唾液酸衍生方法,除至今仍被廣泛使用的全甲基化外,近些年來又相繼發展了以醇類、胺類試劑為核心的方法[16]。已有較為全面的文獻[16,55]總結了這些方法的原理,本文僅從衍生試劑的角度歸納并補充相關內容。
3.1 醇類試劑
在有偶聯劑存在的情況下,甲醇/乙醇能夠與唾液酸的羧基發生酯化反應[56,57]。該反應對連接位置不同的唾液酸具有很高的選擇性。如前文所述,唾液酸經常連接于糖蛋白的末端,且與糖鏈的連接方式也較為多樣化,尤以α2,3-與α2,6-連接最為常見。其中,以α2,6-連接的唾液酸的羧基能夠與醇類衍生試劑發生酯化反應;以α2,3-連接的唾液酸則能夠與相鄰的單糖殘基(通常是半乳糖)發生縮合反應,生成內酯。衍生反應的高選擇性使得糖鏈產生結構差異與相對分子質量差異,有利于在色譜與質譜分析中鑒定唾液酸的連接方式。以甲醇衍生為例,如果糖鏈中存在α2,6-連接的唾液酸,則相對分子質量增加14(見圖4a);如果糖鏈中存在α2,3-連接的唾液酸,則相對分子質量減少18(見圖4b)[57]。此類方法通過相對分子質量差異鑒別不同連接方式的唾液酸,一般稱之為“連接特異性”(linkage-specific)衍生方法。

圖4 唾液酸的連接特異性衍生示意圖
3.2 胺類試劑
與醇類試劑相似,以甲胺、二甲胺、異丙胺等為代表的胺類試劑也可以在無水條件下與α2,3-、α2,6-連接的唾液酸發生特異性反應。其中,α2,6-連接的唾液酸發生羧基酰胺化反應;α2,3-連接的唾液酸發生內酯化反應,通過質譜分析衍生后的糖鏈就可以鑒定唾液酸的連接方式[58,59]。截至目前,已經被用于連接特異性衍生的試劑有10余種,不同試劑的衍生效果存在一些差異性,以異丙胺的特異性最高[59]。值得注意的是,連接特異性的衍生方法主要適用于樣品中僅含有α2,3-與α2,6-連接的唾液酸,而對于α2,8-連接的結構,其能與相鄰唾液酸的羥基發生內酯化反應(見圖4c),模式與α2,3-連接的唾液酸較為相似[60]。
α2,3-連接的唾液酸形成的內酯穩定性不高,連接特異性的衍生方法被進一步優化,通過兩步反應依次衍生α2,6-與α2,3-連接的唾液酸。在第一步反應中,α2,6-連接的唾液酸被酯化或胺化,α2,3-連接的唾液酸被內酯化;在第二步反應中,內酯化的唾液酸開環后被其他胺類試劑繼續衍生[59,61]。兩步反應既保證了衍生產物的特異性,也增強了其穩定性,逐漸成為表征酸性N/O-聚糖的常用方法之一。
胺類試劑的特異性衍生作用通常在1-(3-二甲基氨基丙基)-3-乙基碳二亞胺和1-羥基苯并三唑存在的情況下發生。若改變反應條件則可以消除這種特異性,例如,在1-羥基苯并三唑不存在的情況下,甲胺、甲苯胺可以同時胺化α2,3-、α2,6-和α2,8-連接的唾液酸[60,62,63]
3.3 肼類試劑
與胺類試劑相似,肼類試劑也可以與唾液酸的羧基發生縮合反應,生成酰肼衍生物。事實上,已有多種肼類試劑被用于衍生寡糖的還原端,但用于衍生唾液酸的試劑仍然不多,主要為乙酰肼(acetohydrazide)[40]。另一種肼衍生方法則是由羥基在強氧化劑作用下生成的酮基與肼類試劑發生反應[64]。當氧化劑高碘酸鈉的濃度達到10 mmol/L時,寡糖的末端殘基均可與肼反應;若降低高碘酸鈉的濃度至1 mmol/L,則只有糖鏈末端的唾液酸殘基與肼反應,通過此方法可以選擇性富集唾液酸化的糖肽[15,65]。
4 在糖脂水平上的唾液酸衍生方法
糖脂是一類由寡糖與脂類共價連接而成的糖綴合物,部分糖脂的糖鏈末端含有唾液酸殘基,以神經節苷脂(ganglioside)最為典型。神經節苷脂大量存在于神經系統中,參與眾多生理過程,并在一些疾病中發揮重要作用[6,66]。一般而言,通過質譜的負離子模式可以直接分析唾液酸化的糖脂[67,68],不過,因衍生后的糖脂在提高色譜分離度以及穩定唾液酸殘基等方面具有一些優勢,故衍生化方法在相關研究中也有較為廣泛的應用。
4.1 甲基化試劑
全甲基化方法于20世紀60年代已被用于衍生糖脂,現如今仍然是重要的糖脂衍生方法之一[66,69,70]。常規的全甲基化方法會掩蓋天然分子中的甲基化修飾,研究者采用氘代碘甲烷衍生神經節苷脂,并在藍指海星(Linckialaevigata)中發現了C8位置存在天然甲基化修飾的唾液酸[71]。全甲基化衍生一般需要在強堿性試劑(如氫氧化鈉)存在的情況下發生;若改變反應條件,取消強堿性試劑,則碘甲烷主要與唾液酸的羧基反應生成甲酯類物質[72,73]。類似地,3-甲基-1-對甲苯基三氮(3-methyl-1-p-tolyltriazene)可以代替碘甲烷完成羧基的甲基化反應[74,75]。相對于全甲基化方法而言,甲基化的反應條件較為溫和,有利于抑制唾液酸殘基上的修飾基團被破壞。值得注意的是,甲基化方法主要衍生α2,3-與α2,6-連接的唾液酸,而α2,8-連接的唾液酸易在反應過程中被內酯化[60],通過此現象應當可以特異性地鑒別α2,8-連接的結構。
4.2 胺類試劑
近些年來,胺化反應逐漸被引入到唾液酸化糖脂的色譜與質譜分析中。胺化試劑與羧基發生縮合反應后能夠將酸性基團中性化,有利于穩定唾液酸殘基。糖脂被2-(2-吡啶胺)乙胺(2-(2-pyridilamino)ethylamine)衍生后,一方面能夠產生熒光從而便于通過液相色譜-熒光檢測方法分析;另一方面也可以改變二級質譜的碎片化模式,有利于簡化譜圖。對于烷基胺類試劑而言,其在無水條件下可以衍生唾液酸。而最近一項研究[60]表明,甲胺、乙胺等試劑在水溶液中依然可以高效率衍生唾液酸化糖脂;與之相對,異丙胺、二甲胺等試劑在相同條件下卻幾乎不與羧基反應。連接特異性的衍生方法也適用于糖脂,其中,α2,6-連接的唾液酸發生羧基酰胺化反應;α2,3-與α2,8-連接的唾液酸發生內酯化反應,而內酯化的唾液酸在開環后可以繼續被其他胺類試劑衍生[60]。
在1 mmol/L高碘酸鈉存在的情況下,唾液酸殘基的C7與C8鍵斷開后生成酮基,生成的酮基可被胺類試劑進一步衍生,近期文獻[76]報道了一種基于此操作的同整質量標記(isobaric tag)[77]唾液酸化糖脂的方法。串聯質譜標簽(tandem mass tag)通常被用于標記多肽、N/O-聚糖的還原端[78,79],上述方法則創新性地將其拓展應用于標記糖脂。不過,此方法會破壞唾液酸殘基的結構,導致關鍵的生物學信息被丟失。
5 結論與展望
經過幾十年的發展,研究者們已經建立了多種唾液酸衍生方法,以便增強對其生物功能的認識。但開發新的衍生方法與檢測技術仍然是當前糖生物學領域的重要話題與關鍵任務。
單糖水平上的唾液酸衍生方法應當致力于解決如下幾個問題:第一,減少副反應的發生;第二,消除目標分子的端基異構體等;第三,增強修飾基團的連接位點與同分異構體鑒別能力;第四,發展同位素標記等相對定量分析技術。N/O-聚糖水平上的唾液酸衍生方法相對成熟,當前主要面臨色譜分離度與檢測靈敏度不能滿足需求的問題,以及相關方法沒有得到規范化應用等問題。糖脂水平上的衍生技術則需要考慮:第一,建立能夠改善待測分析物分子的碎片化模式的方法;第二,將N/O-聚糖衍生方法推廣到本領域內;第三,建立溫和的同位素標記方法。
猜你喜歡
中老年保健(2021年9期)2021-08-24 03:52:04
河北畫報(2021年2期)2021-05-25 02:07:46
中學生數理化(高中版.高考理化)(2020年2期)2020-04-21 05:33:04
兒童繪本(2020年5期)2020-04-07 17:46:30
兒童故事畫報(2019年5期)2019-05-26 14:26:14
意林原創版(2016年10期)2016-11-25 10:28:30
Coco薇(2016年2期)2016-03-22 02:42:52
山東青年(2016年1期)2016-02-28 14:25:23
Coco薇(2015年1期)2015-08-13 02:47:34
小雪花·成長指南(2015年7期)2015-08-11 15:03:12