塞來昔布的合成及表征
2019-11-07 11:04:16王福生蘇艷華
生物化工 2019年5期
王福生,蘇艷華
(瑞陽制藥有限公司,山東淄博 256100)
塞來昔布是新一代非甾體鎮痛抗炎藥,通過選擇性抑制環氧化酶-2(COX-2)來抑制前列腺素的合成,從而達到抗炎癥、鎮痛的效果。當前國內外報道塞來昔布的合成文獻[1-3]較多,較為成熟的路線如圖1所示。

圖1 塞來昔布的合成路線
本文在參考相關文獻[4-5]后采用上述路線,并對上述路線的具體工藝進行了改進。本文中化合物4采用乙醇鈉(乙醇鈉溶液含量16%)為縮合劑合成化合物5,合成后的化合物5不經母液分離直接與化合物3反應兩步一鍋生成化合物1,收率達89.5%,較其他文獻[6-8]工藝收率和質量有明顯提高。
1 實驗部分
1.1 儀器及試劑
Vario EL Ⅲ元素分析儀(Elementar公司);Nexus型傅里葉變換紅外(FT-IR)分析儀(Thermo Nicolet公司);Waters SQD2液相色譜質譜聯用儀(Waters公司);Avance Ⅲ600MHz核磁共振譜儀(BRUKER公司)。
對氨基苯磺酰胺(AR)、鹽酸(AR)、亞硝酸鈉(AR)、無水亞硫酸鈉(AR)、對甲基苯乙酮(工業級)、三氟乙酸乙酯(AR)、乙醇鈉溶液(工業級)、三氟乙酸(AR)、異丙醇(AR)、乙醇(AR)。
1.2 塞來昔布的合成
1.2.1 對肼基苯磺酰胺鹽酸鹽(化合物3)的合成
2L三口燒瓶中加入鹽酸(145g,1.45mol)、對氨基苯磺酰胺(100g,0.58mol)和去離子水300g,攪拌降溫至0℃。將配制好的亞硝酸鈉溶液(44.07g亞硝酸鈉溶于126g水中)緩慢滴加到三口燒瓶中,控溫0~5℃,約30 min滴畢,滴完后保溫攪拌10 min。
5L三口燒瓶中加入水330g,在攪拌下加入無水亞硫酸鈉(182.98g,1.45mol),降溫至0℃,再加入碎冰130g,將上述反應液加入此體系中,用水20g沖洗2L三口燒瓶,操作完畢后保溫攪拌10 min。然后水浴升溫至80℃,加入鹽酸(392.97g,3.93mol),溶解澄清后停止攪拌和加熱,自然降溫至室溫后析晶12 h,然后抽濾干燥得到中間體對肼基苯磺酰胺鹽酸鹽約102.41g,純度98.92%,收率為78.84%。
1.2.2 塞來昔布(化合物1)的合成
2L三口燒瓶中加入乙醇鈉溶液(230.84g,0.56mol),在攪拌下加入三氟乙酸乙酯(80.458g,0.57mol),攪拌5 min后加入對甲基苯乙酮(57.13g,0.43mol)。
水浴加熱升溫至50℃,保溫攪拌2 h,反應液直接用于下一步反應。
5L三口燒瓶中加入對肼基苯磺酰胺鹽酸鹽(100g,0.45mol)、乙醇221g和水157.59g,攪拌下加入三氟乙酸(50.98g,0.45mol),水浴加熱升溫。當溫度升至50℃時,將上步反應液滴加到此體系中,然后保溫攪拌30 min,再加入水203.39g,升溫至65℃,再滴加水191.67g,保溫攪拌30 min,停止加熱,攪拌析晶約3 h后抽濾,使用50%乙醇充分洗滌2次后干燥得到固體約145.3g,純度99.12%,收率為89.5%。
重結晶:500mL三口燒瓶中,裝有攪拌、溫度計和冷凝管,將50.0g塞來昔布粗品加入三口燒瓶中,再加入160g異丙醇,加熱溶解澄清后,再加入2g活性炭,回流攪拌20min后濾除活性炭,將濾液轉至干凈反應瓶中,加熱回流澄清后,緩慢加入8.0g水,緩慢降溫析晶待降至20~25℃后,再析晶6h后抽濾,使用少量冷異丙醇的沖洗后干燥得45.2g,純度為99.83%,收率為90.4%。
2 結果及討論
2.1 塞來昔布的元素分析
在制備塞來昔布后,為了進一步確定其結構,對產物進行了C、H、N的元素分析,其結果如表1所示。
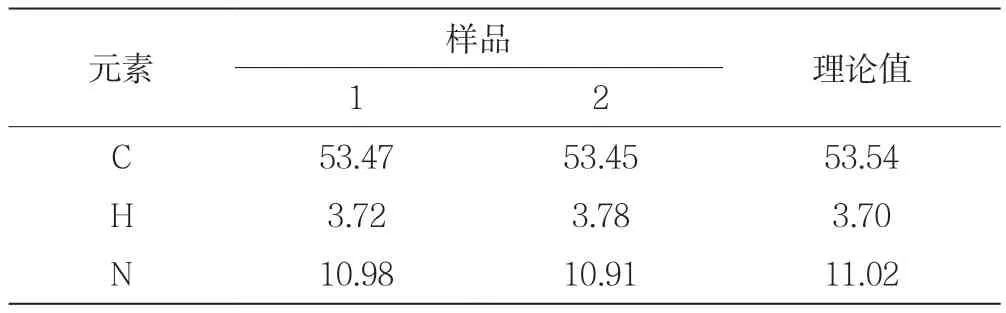
表1 塞來昔布的元素分析
由表1可知,根據樣品中C、H和N元素分析結果,各元素實測值平行性良好,實測值與理論值基本一致,為進一步確定其結構進行了IR分析。
2.2 塞來昔布的IR分析
將制備的塞來昔布進行紅外光譜分析,其主要吸收峰及歸屬如表2所示。

表2 塞來昔布的IR譜各吸收峰及歸屬
由表2可以看出:(1)3098cm-1為苯環中=C-H的伸縮振動吸收,1563cm-1、1499cm-1為苯環骨架的伸縮振動吸收,846cm-1為苯環中=C-H的彎曲振動吸收,說明結構中含有苯環結構;(2)1375cm-1為甲基的C-H彎曲振動吸收,說明結構中含有甲基結構;(3)3339cm-1、3233cm-1為磺酰胺的N-H伸縮振動吸收,1348cm-1為磺酰胺基的SO2的伸縮振動吸收,說明結構中含有磺酰胺結構。
解析結果表明:該樣品的紅外光譜圖特征與目標化合物的化學結構基本相符合。
2.3 塞來昔布LC/MS分析
將制備的塞來昔布進行質譜分析,其分子離子峰及歸屬如表3所示。

表3 塞來昔布的分子離子峰及歸屬
由表3可知,質譜測得本品的分子離子峰[M+H]+,其質荷比m/z為382.1,與塞來昔布的分子離子峰(分子量為381.37)一致。
2.4 塞來昔布的1H-NMR分析
將制備的塞來昔布樣品進行1H-NMR譜檢測,1H-NMR((CD3)2SO,600MHz):δ7.906(d,2H,J=8.4Hz,Ar-H),7.561(s,2H,NH2),7.550(d,2H,Ar-H),7.227(d,2H,Ar-H),7.222(d,2H,Ar-H),7.190(s,1H,吡唑環CH),2.324(s,3H,CH3),綜合以上信息,與塞來昔布結構信息吻合。
3 結論
(1)通過元素分析、紅外光譜法、質譜法和1H-NMR的檢測和表征,確認了塞來昔布目標產物的合成。
(2)本文中化合物4采用乙醇鈉(乙醇鈉溶液含量16%)為縮合劑合成化合物5,合成后的化合物5不經處理分離直接與化合物3反應兩步一鍋生成化合物1,收率達89.5%,經精制后純度達99.83%,該路線總收率為63.8%,經改進后其收率和質量有明顯提高。
猜你喜歡
哲學評論(2021年2期)2021-08-22 01:53:34
民用飛機設計與研究(2020年4期)2021-01-21 09:15:02
中華詩詞(2019年7期)2019-11-25 01:43:04
模具制造(2019年3期)2019-06-06 02:10:54
電子制作(2018年18期)2018-11-14 01:48:24
山東工業技術(2016年15期)2016-12-01 05:31:22
影視與戲劇評論(2016年0期)2016-11-23 05:26:01
現代企業(2015年9期)2015-02-28 18:56:50
中國中醫藥現代遠程教育(2014年11期)2014-08-08 13:23:44
終身教育研究(2014年5期)2014-02-28 01:23:06
