HBV表面抗原preS1激活Wnt信號通路導致肝癌發生的機制研究
2019-11-05 07:14:54李云靜譚華炳劉志新邱雪梅
中國人獸共患病學報 2019年10期
關鍵詞:肝癌
袁 杰,熊 希,李云靜,譚華炳,劉志新,3,王 婭,邱雪梅,李 蓓,楊 靖,
乙型肝炎病毒(Hepatitis B Virus, HBV)屬嗜肝性DNA病毒科,其基因組由不完全閉合環狀雙鏈DNA(Relaxed circular DNA,rcDNA)組成,長約3 200對堿基[1]。HBV感染分急性和慢性感染,全球約有20億人感染過HBV,其中大約3.5億是慢性感染者。每年死于慢性乙肝(Chronic hepatitis B,CHB)引起的肝纖維化(Liver fibrosis)、肝硬化(Liver cirrhosis)及肝細胞癌(Hepatocellular carcinoma,HCC)的人數約有78萬[2]。我國是HBV感染大國,不僅HBV攜帶者基數大(約1.2億左右),而且HBV攜帶者極易轉化為HCC患者,每年導致大量的死亡病例[3]。因此,研究HBV感染導致HCC的分子機制對于防治病毒感染至關重要。
HBV感染與肝癌發生具有正相關關系[4],但是學術界對于HBV病毒如何導致肝細胞癌化目前并沒有一致結論。目前的文獻資料關于此方面的闡述主要集中在3個方面:① HBV 基因組整合到宿主基因組的關鍵位置導致宿主基因組抑癌基因的失活或者是原癌基因基因的活化[5-7];② HBV感染細胞造成宿主細胞的炎癥反應,進而激活下游信號通路,導致肝細胞癌化[8-9];③ HBV進入細胞之后病毒 DNA編碼的結構蛋白、分泌蛋白, 或者通過表觀遺傳導致宿主基因突變和染色體重排等[10-12]。
HBV在細胞中有2種存在方式,即游離型和整合型。HBV陽性的肝細胞癌患者中幾乎均可檢測到HBV基因組整合[13]。HBV 基因整合至宿主基因組中有利于其持久感染, 長期的慢性炎癥可導致肝細胞生命周期改變和增殖, 這增加了宿主基因組DNA終端的數量, 從而有利于HBV基因組整合到宿主基因組中。在有些案例中,HBV基因組整合與肝癌發生具有一定的相關性,但是研究者仍需要更加堅實的臨床高通量測序數據來進行深入分析[14]。
檢測急性和慢性HBV感染者可在外周血單個核細胞(Peripheral Blood Mononuclear Cells,PBMC)中檢測到游離和整合的病毒基因組[15]。Shi等[16]曾報道HBV基因組特定位點的整合可導致T細胞缺陷。HBV除引起肝臟細胞的炎癥反應之外,亦可以引發外周血淋巴細胞持續性的炎癥反應。但是,目前仍未見報道HBV感染與血液系統的癌癥發生具有相關性。
HBV基因組進入肝細胞中之后,自身也可以表達一系列的蛋白。雖然經過幾十年的研究,很多蛋白的功能仍未能得到完整的闡釋,但有證據顯示,HBV DNA表達合成成熟的preS/S和X蛋白觸發級聯信號可導致肝細胞惡變[17]。
近期,有研究報道HBV的表面膜蛋白preS1可促使正常肝細胞向癌化肝細胞發展[18]。在已有研究結果的基礎上,我們進一步研究發現preS1通過抑制β-catenin的磷酸化激活Wnt信號通路,進而促進正常肝細胞向癌化肝細胞轉化。本研究旨在能為HBV感染與肝癌發生的機制研究提供新的思路和線索,為未來臨床基因診斷和治療肝癌奠定理論依據。
1 材料和方法
1.1臨床組織標本 12例肝癌患者的石蠟包埋肝臟腫瘤組織標本來源于湖北醫藥學院附屬人民醫院病理科。其中,6 例標本是HBsAg陽性,6 例標本是HBsAg陰性。肝臟腫瘤組織經4%甲醛溶液固定,修塊、脫水、浸蠟、包埋、切片,免疫組化染色,光學顯微鏡下觀察腫瘤切片情況,并拍照記錄。所有的人體組織材料的使用均通過湖北醫藥學院倫理審查委員會批準。
1.2細胞培養 L02肝臟細胞系,HepG2肝癌細胞系和HEK293T細胞系購自中國典型培養物保存中心(China center for Type Culture Collection,CCTCC),用含10%胎牛血清(Gibco Life Technologies, USA)、雙抗(青霉素100 U/mL,鏈霉素100 mg/mL)(Gibco Life Technologies, USA)的DMEM高糖培養基(Gibco Life Technologies, USA),于 5% CO2的恒溫培養箱中37℃培養。
1.3載體構建和引物合成 慢病毒載體pWPXLD (Addgene plasmid # 12258) 、pLKO.1-TRC (Addgene plasmid # 10878)及輔助質粒均購自Addgene公司。preS1基因全長使用PCR擴增后,連接入pWPXLD載體的多酶切位點PmeI(NEB, #R0560S)和BamHI(NEB, #R0136S)之間。根據文獻[18],一段preS1 shRNAs 序列構建入pLKO.1-TRC載體的AgeI(NEB, #R0552S)和EcoRI(NEB #R0101S)酶切位點之間。所有的構建過程均參照載體說明書進行。preS1基因PCR引物序列和shRNA序列均為華大基因合成,具體序列信息見表1。
表1 引物序列信息
Tab.1 Information of primer

引物名稱引物序列(5′-3′)退火溫度/℃引物用途preS1 FAGCTTTGTTTAAACATGGGAGGTTGGTCTTCC62載體構建preS1 RGCGGGATCCCTTGTCATCATCGTCCTTG-TAGTCGGCCTGAGGATGACTGTCTC62載體構建preS1 shRNA FCCGGGATTCTTTCCCGATCACCAGTTG-GACTCGAGTCCAACTGGTGATCGGGAAAGAATCTTTTTG—載體構建preS1 shRNA RAATTCAAAAAGATTCTTTCCCGATCAC-CAGTTGGACTCGAGTCCAACTGGTGATCGGGAAAGAATC—載體構建ALDH1 FAAAGAAGCTGCCGGGAAAAG58QRT-PCRALDH1 RCCCCATGGTGTGCAAATTCA58QRT-PCROCT4 FAGAACATGTGTAAGCTGCGG58QRT-PCROCT4 RGGTTCGCTTTCTCTTTCGGG58QRT-PCRSOX2 FAGCTCGCAGACCTACATGAA58QRT-PCRSOX2 RTGGAGTGGGAGGAAGAGGTA58QRT-PCRFGF20 FTTAGAGGTGTGGACAGTGGT58QRT-PCRFGF20 RAGTGCCACAAAATACCTGCG58QRT-PCRDKK1 FAGGTTCTGTTTGTCTCCGGT58QRT-PCRDKK1 RCTCCACAGTAACAACGCTGG58QRT-PCRWISP1 FGACTTTACCCCAGCTCCACT58QRT-PCRWISP1 RGTAGTCACAGTAGAGGCCCC58QRT-PCRCCND1 FGCATGTTCGTGGCCTCTAAG58QRT-PCRCCND1 RCGTGTTTGCGGATGATCTGT58QRT-PCRGAPDH FTCGTGGAAGGACTCATGACC58QRT-PCRGAPDH RATGATGTTCTGGAGAGCCCC58QRT-PCR
1.4慢病毒包裝和感染 構建完成的慢病毒質粒使用293T細胞系進行包裝。每個6 cm的培養皿,使用Lipofectamine? 2000 (Thermo Fisher Scientific, USA) 轉染12 mg的目的質粒,加上6 mg的輔助質粒pMD2G,12 mg的輔助質粒psPAX2。轉染后的48 h和72 h收集細胞培養物上清,并使用0.45 μm的微孔濾膜過濾、分裝后,于-80 ℃超低溫冰箱備用。培養的L02和HepG2細胞系使用包裝好的慢病毒進行感染,并使用流式細胞術分選或是嘌呤霉素篩選陽性細胞。
1.5免疫印跡分析 使用加入了蛋白酶抑制劑的(complete protease inhibitor cocktail, Roche, USA) RIPA裂解液裂解收集的細胞。使用Bradford蛋白定量試劑盒 (Bio-Rad, USA)對提取的總蛋白溶液進行定量,并使用SDS-PAGE進行電泳分離,并轉至NC膜(Nitrocellulose Membrane)。與相應的一抗孵育之前,NC使用含5%脫脂奶粉和0.1%的吐溫20的PBS溶液進行封閉。使用Bio-Rad凝膠圖像成像儀顯影,并保存實驗結果。
1.6實時熒光定量PCR分析 用TRIZOL試劑提取細胞總RNA,用TaKaRa逆轉錄試劑盒將RNA逆轉錄成為cDNA,具體步驟按照產品說明書進行。QRT-PCR檢測使用BIO-RAD CFX96 和 SYBR RT-PCR kits (ROCHE, USA)熒光定量試劑盒進行。反應體系為:10 μL SYBR Green PCR master mix,5 μL稀釋10倍的 cDNA模板和5 μL稀釋10倍的正反引物混合物。反應程序為:95 ℃,3 min;95 ℃,20 s,58 ℃,20 s,72 ℃,20 s,采集熒光信號,運行45個循環;65 ℃至95 ℃繪制溶解曲線。QRT-PCR所使用的引物使用Primer Premier 5.0軟件進行設計,具體所使用引物序列請見表1。
1.7免疫共沉淀 穩定過表達帶FLAG標簽preS1蛋白的L02-preS1細胞系使用加入了蛋白酶抑制劑的(complete protease inhibitor cocktail, Roche, USA)RIPA裂解液冰上裂解1 h。細胞裂解產物高速離心(15 000 g, 15 min, 4 ℃)去除未溶解的細胞碎片。收集的上清加入anti-FLAG標簽抗體(1 μg/mL)4 ℃孵育過夜,然后再加入30 μL的可以吸附IgG的proteinA瓊脂糖珠(CST,#9863)4 ℃孵育2 h。使用RIPA溶液漂洗5~7次。然后高溫洗脫吸附的蛋白,使用SDS-PAGE或是Western-Blot進行檢測。
1.8數據處理 采用SPSS 17.0軟件進行統計學處理。兩兩檢驗采用LSD-t檢驗。以P<0.05為差異有統計學意義。
2 結 果
2.1HBV促進肝癌發生的臨床肝癌樣本分析 通過對 6 例HBV陽性的肝癌標本和 6 例HBV陰性的肝癌樣本進行石蠟切片和免疫組化染色分析,我們發現HBV攜帶者CD133陽性的肝癌干細胞數量要多于對照組(圖1)。基于對臨床樣本的分析,我們推測HBV可以表達某些因子促進肝癌干細胞的產生。
2.2preS1促進肝癌發生的細胞模型分析 近期,已有研究報道preS1可以促進正常肝臟細胞向癌化肝臟細胞轉化[18]。為了進一步深入研究preS1促進肝細胞癌化的具體機制,我們利用慢病毒轉染技術構建了穩定過表達preS1的肝臟細胞模型。與對照組L02-GFP相比,L02-preS1細胞中癌癥細胞相關基因ALDH1、Oct4和Sox2在mRNA水平上調表達(圖2A)。在細胞模型HepG2-preS1中,我們可以得到類似的結果(圖2B)。Western-Blot結果顯示,L02-preS1和HepG2-preS1中ALDH1、Oct4和Sox2的表達水平均因為preS1的過表達而上調(圖2C)。以上結果證實,在肝細胞中過表達preS1可以促進癌癥相關因子的上調表達。
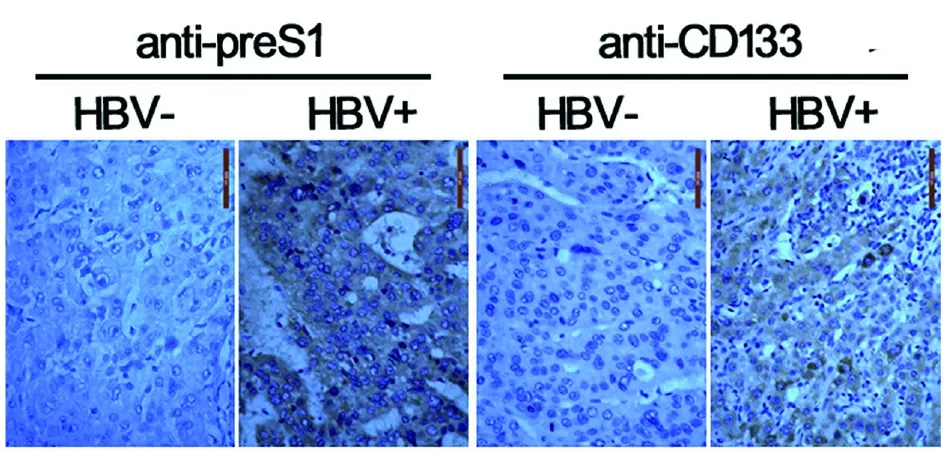
圖1 肝癌病理切片免疫組化染色的顯微照相圖片Fig.1 A micrographic picture of immunohistochemical staining of the pathological section of liver cancer

Graphs show means±SD, n= 3;*: P<0.05; **: P<0.01; ***: P<0.001.圖2 在肝細胞系L02和肝癌細胞系HepG2中穩定過表達preS1可上調表達腫瘤標志分子ALDH1、Oct4和Sox2Fig.2 Stable over-expression of preS1 in hepatocyte line L02 and hepatoma cell line HepG2 can up regulate the expression of tumor markers ALDH1, Oct4 and Sox2
2.3preS1促進肝癌發生的內在機制分析 雖然已有報道preS1促進肝癌產生,但是研究者對其內在機制仍缺乏了解。我們通過免疫共沉淀技術獲取了能與preS1相互作用的蛋白。免疫共沉淀產物經過SDS-PAGE電泳分離,主要條帶經質譜分析,發現preS1與β-catenin存在相互作用(圖3A)。我們進一步對preS1穩定過表達和下調表達細胞模型進行QRT-PCR分析。令人意外的是,在過表達和下調表達preS1之后,在mRNA水平并未檢測到β-catenin的表達變化(圖3B)。我們進一步使用Western-Blot分析β-catenin磷酸化水平的變化。結果顯示:過表達preS1之后,磷酸化的β-catenin表達量降低,而非磷酸化的β-catenin表達量升高;而使用RNA干擾下調preS1的表達量,β-catenin的磷酸化水平可以得到恢復(圖3C)。為了進一步證實上述結果,我們使用不同濃度梯度preS1過表達質粒(0、0.2、0.4 μg/mL)轉染L02細胞,并在轉染48 h 之后檢測磷酸化β-catenin和非磷酸化β-catenin表達量。結果顯示:隨著preS1過表達質粒轉染濃度的增加,磷酸化β-catenin的表達量逐步降低,而非磷酸化的β-catenin表達量逐步升高 (圖3D)。FGF20、DKK1、WISP1和CCND1是Wnt信號通路中可以被β-catenin激活的關鍵下游基因[22]。我們結果顯示:與對照組(L02-GFP)相比,preS1穩定過表達肝細胞系(L02-preS1)在mRNA水平上調表達FGF20、WISP1和CCND1(圖3E)。以上結果說明,preS1可以通過抑制β-catenin磷酸化,進而提高細胞內未磷酸化β-catenin的濃度,從而激活Wnt信號通路。
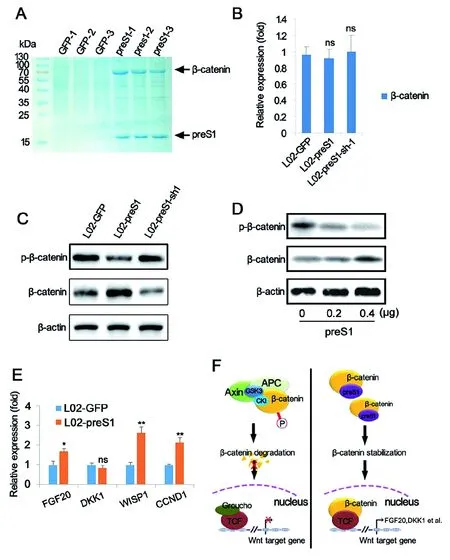
Graphs show means±SD, n=3;ns: no significant difference; *: P<0.05; **: P< 0.01.圖3 preS1通過結合β-catenin,抑制β-catenin磷酸化,從而激活Wnt信號通路 Fig.3 preS1 activates Wnt signaling pathway by binding to β-catenin and inhibiting phosphorylation of β-catenin
3 討 論
CD133作為肝癌干細胞的表面標志物已得到研究者的公認[19]。ALDH1、Sox2和Oct4作為細胞內部表達的關鍵腫瘤干細胞標志分子,也廣泛用于肝癌干細胞的篩選和標記[20]。我們對臨床肝癌標本進行免疫組化分析發現,HBV攜帶者病理切片標本中CD133陽性細胞數量要多于未感染HBV的對照組(圖1)。我們利用慢病毒技術,構建了穩定過表達HBV表面膜蛋白preS1的肝細胞模型。與對照組相比,過表達preS1的肝細胞模型中ALDH1,Sox2和Oct4在mRNA水平和蛋白水平均得到了上調表達(圖2)。相對于肝癌細胞模型HepG2,在正常肝細胞模型L02中過表達preS1,肝癌細胞腫瘤標志物的表達水平上調更加明顯。
已有報道顯示,preS1的表達對于正常肝細胞向癌化肝細胞的轉化具有重要作用[18],但是肝癌發生過程中與preS1相互作用的下游因子及信號通路仍未見報道。利用建立的L02-preS1穩定過表達細胞系,結合免疫共沉淀技術我們獲取了能與preS1相互作用的蛋白。使用SDS-PAGE電泳分離之后,對主要條帶進行質譜分析后發現,preS1在蛋白層面可以和β-catenin相互作用(圖3A)。我們利用慢病毒RNAi載體pLKO.1-TCR,在過表達preS1的L02細胞系中下調表達preS1。令我們意外的是,下調表達preS1,β-catenin在mRNA水平的表達量并沒有變化(圖3B)。隨后,我們使用Western-Blot對β-catenin的蛋白水平進行了檢測。我們發現在L02肝細胞模型中過表達preS1,β-catenin的磷酸化水平受到抑制。而下調preS1的表達量,β-catenin的磷酸化水平可以得到恢復(圖3C)。通過不同濃度梯度的preS1過表達質粒轉染肝細胞系L02,我們進一步證實了preS1的表達可以抑制β-catenin的磷酸化,上調非磷酸化β-catenin的表達水平(圖3D)。β-catenin作為Wnt信號通路上的關鍵蛋白,對于癌癥干細胞的發育至關重要。在經典的Wnt信號通路中,APC、Axin、GSK3和CKI可組成復合物,降解磷酸化的β-catenin,阻止β-catenin和轉錄因子TCF結合,抑制Wnt信號通路開啟[21]。β-catenin和轉錄因子TCF結合會激活下游基因(如FGF20,DKK1,WISP1,CCND1等)的表達[22],從而開啟Wnt信號通路。本研究也證實過表達preS1可以抑制β-catenin的磷酸化,激活FGF20,WISP1和CCND1的表達(圖3E)。
結合已發表的研究結果[18],并對本次的研究結果進行分析之后,我們提出如下觀點:preS1與β-catenin的結合可抑制β-catenin的磷酸化,未磷酸化的穩態β-catenin在細胞內積聚,與下游TCF轉錄因子結合,激活Wnt信號通路,并促進肝細胞癌化(圖3F)。本研究旨在能夠促進我們對HBV導致的肝癌的機制理解,并未將來防治HBV引起的肝癌奠定理論基礎。
利益沖突:無
猜你喜歡
天津醫科大學學報(2019年3期)2019-08-13 06:53:08
中成藥(2016年8期)2016-05-17 06:08:14
癌癥進展(2016年12期)2016-03-20 13:16:17
罕少疾病雜志(2016年5期)2016-03-11 16:34:44
吉林大學學報(醫學版)(2015年1期)2015-12-17 07:47:28
腫瘤預防與治療(2015年1期)2015-09-26 07:26:20
中國當代醫藥(2015年16期)2015-03-01 02:03:11
中國醫藥導報(2015年26期)2015-02-28 22:07:59
肝膽胰外科雜志(2015年4期)2015-02-27 11:12:34
肝膽胰外科雜志(2015年4期)2015-02-27 11:12:24
