鈣鈦礦NaFeF3結構物性的理論研究及應力和摻雜調控
2019-10-23 01:22:32宋蕊馮凱林上金何曼麗仝亮
物理學報 2019年14期
關鍵詞:結構
宋蕊 馮凱 林上金 何曼麗 仝亮
(陸軍工程大學基礎部,南京 211101)
利用第一性原理計算研究了氟化物鈣鈦礦NaFeF3的基態電子態和磁構型,并模擬分析了襯底應力和陰離子置換對材料基本物性的影響.計算結果表明,NaFeF3塊材的G-AFM磁基態十分穩定,不會受到襯底應力調控以及F位氧摻雜的影響.當低濃度摻雜(~8.3%)時,氧離子傾向于替換FeF2面內的F,形成非對稱的Fe-O-Fe鏈,導致產生局域極化.此外,不對等的Fe—O鍵還會在氧離子兩側的Fe上形成電荷序,進而產生非零凈磁矩.值得注意的是,該局域極化和亞鐵磁性均源于不等位氧摻雜,因此有可能通過外電場調控實現極化翻轉并同時改變凈磁矩的方向實現電控磁.更高濃度的氧摻雜會使得Fe的化合價整體升高,Fe離子上的局部電荷序和凈磁矩隨之消失.該研究結果有望促進氟化物的理論和實驗研究,并為多鐵性材料的設計研發提供新的材料.
1 引 言
過渡金屬氧化物鈣鈦礦材料ABO3具有特殊的氧八面體構型,加之B位過渡元素豐富的物理特性,使得該類材料在電學、磁學以及結構方面表現出異常豐富的物理性質,如巨(龐)磁電阻效應、超導電性、多鐵電性[1-4]等.上述這些特殊性質都是由材料內部的多種自由度相互耦合與競爭導致的.該類材料的這種強關聯特性使其成為了凝聚態物理和強關聯電子體系的理想研究平臺.
與氧化物鈣鈦礦材料ABO3相類似,氟化物鈣鈦礦材料ABF3也具有眾多特殊性質,針對此類材料的研究自20世紀60年代以來未曾停止過.例如在鈣鈦礦型KBF3系列材料中,KCrF3擁有與錳氧化物相類似的巨磁電阻效應[5]; KCuF3是自旋1/2海森伯反鐵磁體,其奈爾溫度接近室溫(280 K)[6];KFeF3中電荷有序導致存在多鐵電性[7].即便如此,與氧化物鈣鈦礦材料相比,氟化物鈣鈦礦材料的研究還是相對滯后,相關報道并不多見[8,9].近年來,隨著NaBF3系列材料在光催化活性和電化學性質方面的深入研究,該類氟化物作為環保類功能材料以及電極材料獲得了一定的關注[10-13].
目前,關于NaFeF3材料電學、磁學及結構信息的詳盡報道仍然較少.與立方相KFeF3相比,由于A位Na離子半徑較小,材料中氟八面體必將隨之發生畸變,相關實驗推斷其結構對稱性屬于Pnma空間群.基于目前人們對于鈣鈦礦結構的了解,結合在材料物性分析領域廣泛應用的第一性原理計算方法,本文針對鈣鈦礦結構NaFeF3的電磁學性質展開系統研究,分析應力以及陰離子替換對材料電子結構的影響,希望能夠為后續實驗提供參考.
2 理論模型和計算方法
2.1 理論模型
本文計算采用NaFeF3正交相鈣鈦礦結構,該結構屬于Pnma空間群.其中FeF6八面體彼此間共角相連構成三維框架,Na離子位于FeF6八面體圍成的空間中心.每個結構單胞中包含4個分子式,其具體結構如圖1所示.相關實驗報道的晶格結構參數為 a=5.48 ?,b=5.66 ?,c=7.88 ?[14].
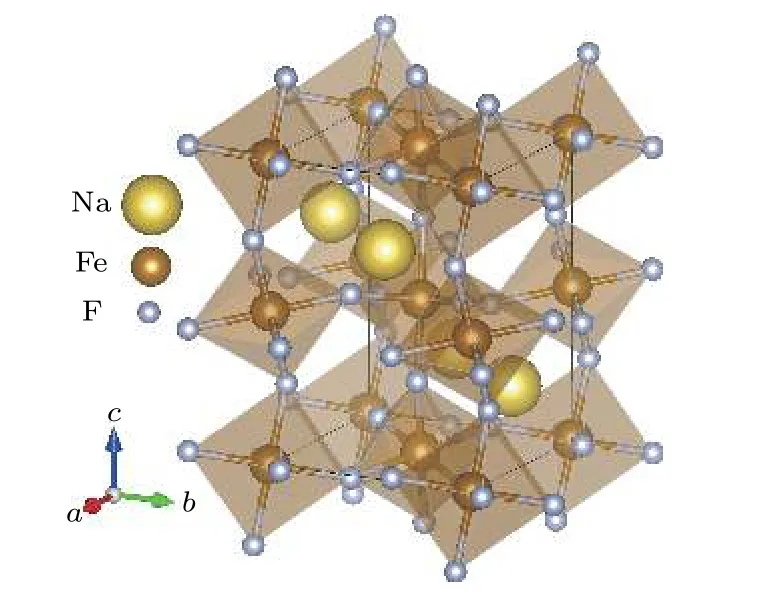
圖1 鈣鈦礦型氟化物NaFeF3的結構示意圖 淺褐色的是由氟離子構成的FeF6八面體,其中心為B位Fe離子,近鄰八面體之間通過共角F離子相連,A位Na離子位于FeF6八面體圍成的三維框架中Fig.1.Schematic diagramof perovskite fluoride NaFeF3.The light brown structures are corner-sharing FeF6 octahedra.The center of octahedra are B-site iron ions,which are shown in dark brown.The yellow Na ions are located in the framework surrounded by the FeF6 octahedra.
2.2 計算方法
文中計算采用基于密度泛函理論(DFT)的第一性原理計算軟件(Vienna ab initio simulation package,VASP)完成[15,16].具體計算采用廣義梯度近似 (GGA)[17]的 Perdew-Burke-Ernzerhof for solids (PBEsol)函數對電子與電子之間的交換關聯作用進行處理[18].體系中Na,Fe,F原子外層價電子組態分別為2p63s1,3d64s2,2s22p5.在布里淵區內以Gamma點為中心采用Monkorst-Park法[19]取5 × 5 × 3的K點網格進行積分求和,平面波截斷能設為500 eV.晶體結構幾何優化迭代的能量收斂判據為1 × 10—5eV,原子力收斂判據為0.01 eV/?.考慮到Fe元素3d電子的強關聯特性,計算中采用Hubbard模型加入on-site庫侖排斥能U進行擬合修正.根據文獻[20]及實際驗算,當Fe的3d軌道上的有效修正U取4 eV時,優化后的結構參數以及磁性基態與實驗吻合較好,文中后續計算均以該U值進行.
3 計算結果與分析
3.1 結構性質
NaFeF3塊材全局優化后的結構對稱性屬于Pnma空間群,優化所得晶格參數a=5.47 ?,b=5.67 ?和c=7.78 ?,與實驗報道值5.48 ?,5.66 ?,7.88 ?均吻合較好.優化后結構沿[110]和[001]方向 Fe—F—Fe鍵角分別為 145.5°和 143.0°.可見相對于立方相KFeF3而言,NaFeF3中Na離子半徑減小導致結構產生顯著的GdFeO3型畸變.該畸變主要表現為FeF6八面體的扭轉,用Glazer標記法可表述為 a—a—c+模式,具體又分為 a—a—c0和 a0a0c+兩種模式,即沿著贗立方的[100]和[010]方向FeF6八面體反相等幅轉動,而在[001]方向則為同相轉動.此外,Fe—F鍵長也出現分化,表明FeF6八面體在扭轉的同時還發生了Jahn-Teller畸變.理論計算與相關實驗數據詳見表1.從表1中不難看出,本文理論計算結果與實驗報道值較為吻合,進一步驗證了相關計算參數的合理性.
3.2 磁性及電學性質
為了研究NaFeF3的電磁學性質,首先對材料的磁基態進行了計算.計算中考慮了四種可能的磁序: 1)鐵磁性(FM),即ab面內及面間自旋取向均為平行排列; 2)A型反鐵磁(A-AFM),即ab面內自旋平行排列,面間呈反平行排列; 3)C型反鐵磁(C-AFM),即ab面內反鐵磁排列,面間最近鄰自旋間平行排列; 4)G型反鐵磁(G-AFM),即ab面內和面間最近鄰自旋間均反平行排列.通過比較不同磁序間優化后的最終體系能量,發現NaFeF3的磁性基態為G-AFM,該結果與實驗報道一致[14].此外,計算結果顯示每個Fe2+離子上的平均磁矩為3.8μB,與Fe2+的3d6的高自旋態S=2相一致.

表1 NaFeF3晶格優化后的結構參數與相應實驗報道值Table 1.Optimized structural parameters of NaFeF3 and the corresponding experimental values.
我們注意到已有文獻報道NaMF3(M=Mn,Co,Ni)為弱鐵磁,即在G型反鐵磁的基礎上,自旋略有傾斜從而導致產生微小的非零磁矩[21-23].但目前NaFeF3相關實驗報道較少,材料的精細磁結構性還有待中子散射等實驗的進一步驗證.考慮到復雜磁性計算需要構建較大的結構體系,涉及原子數目眾多,對計算資源和時間都有極高要求,實際計算不具有可操作性.而目前材料磁基態又與現有實驗報道基本吻合,因此除了磁性基態對比驗算外,后續計算分析均是在G-AFM磁序下進行的.
圖2是NaFeF3塊材的分波態密度圖,不難看出,材料能隙為3.0 eV,經過雜化泛函(HSE)驗算,塊材帶隙為3.14 eV,進一步確認了計算中所用參數的合理性和結果的可信度.費米面附近電子態主要來自Fe和F的貢獻,Na的態密度分布主要局域在相對較深層能級.具體而言,在價帶頂和導帶底處態密度主要源于Fe的3d軌道貢獻.此外在—2.4—1.7 eV的能量范圍內,Fe的3d和F的2p軌道存在較強的雜化,而Na的2p軌道則分布于—3 eV以下能量區域.考慮到Fe—F鍵中陰陽離子較大的電負性差異,不難理解FeF6八面體中Fe—F離子鍵作用較強,—2 eV及以下深能級Fe 3d和F 2p軌道雜化較強,而費米面處材料的電學性質主要由Fe離子3d軌道決定.此外,費米面附近相對獨立且展寬較窄的態密度分布可以近似看作是FeF6的分子軌道.
值得一提的是NaFeF3的能隙寬度與TiO2的相近,相應的吸收波長主要位于400 nm附近的紫外波段.類似于TiO2的能帶調制,想要提高材料的光電轉化效率,最直接的方法之一就是通過元素摻雜置換.后續我們會對材料進行硫族陰離子置換,研究其對材料能帶以及能隙的影響.
3.3 應力對磁性和電學性質的影響
由于強關聯電子體系中晶格和自旋、軌道、電子之間存在較強的耦合作用,襯底應力對材料結構、磁性和電學性質有著非常豐富的調制作用,例如在襯底作用下材料會表現出塊材母相中不具備的磁序或鐵電性等.NaFeF3由于GdFeO3形畸變以及Jahn-Teller效應導致晶格常數a,b不再相等,通過施加雙軸應力,是否會誘導出新的不同于塊材的磁性? 為此,我們對材料進行了一系列雙軸應力下的磁性模擬計算.結果如圖3所示,在具有現實可行性的 ± 5%應力范圍內,G-AFM始終是能量最低的磁基態,NaFeF3并未出現任何新的磁序,母相G-AFM十分穩定.Fe2+離子上的磁矩也始終保持在3.8μB左右,并未隨應力改變出現明顯變化.此外,材料的晶格結構對稱性也并未出現改變.
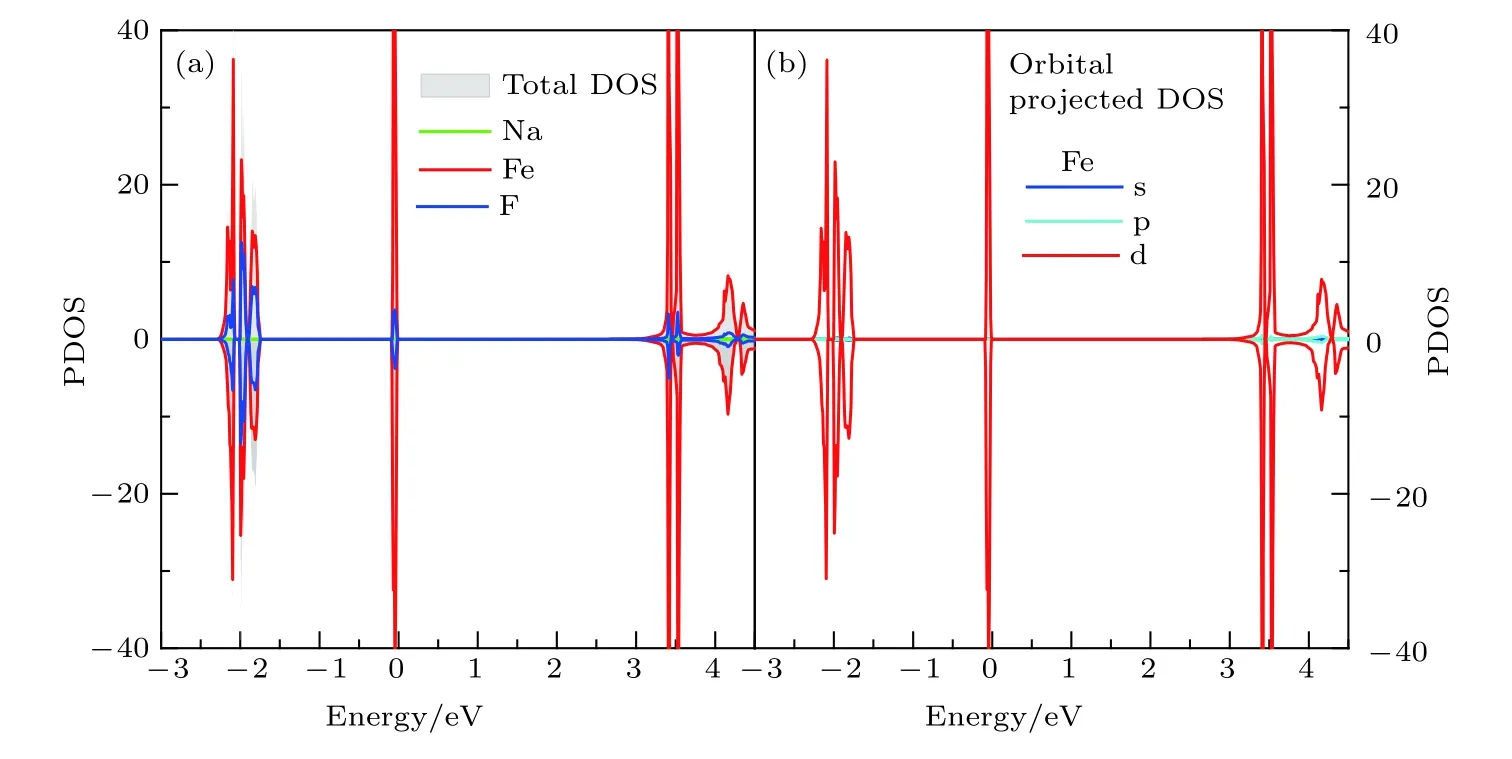
圖2 (a)NaFeF3塊材的原子分波態密度圖; (b)塊材中Fe2+離子的3d軌道分波態密度Fig.2.(a)The projected DOS of bulk NaFeF3; (b)the 3d orbital-projected DOS of Fe2+ ion.
該G-AFM磁基態的成因可能源于Fe2+離子的3d6高自旋電子構型,eg軌道電子通過F-離子的2p軌道與近鄰Fe2+離子形成反鐵磁超交換作用.上述計算結果表明該超交換作用較強,在應力可調控范圍內,NaFeF3的結構及磁性較為魯棒,并未受到明顯影響.
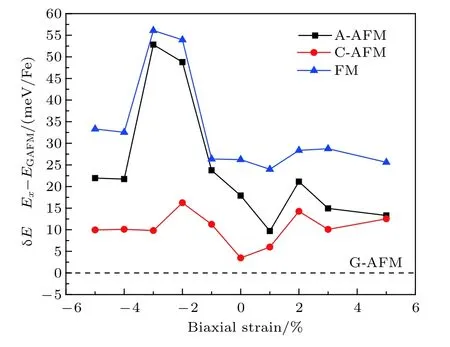
圖3 面內雙軸應力對NaFeF3塊材中A-AFM,C-AFM,FM磁序以及G-AFM磁序之間相互能量差的影響,以GAFM能量作為參考值,如虛線所示Fig.3.The biaxial strain dependent energy differences between A-AFM,C-AFM,G-AFM and FM states.The energy of G-AFM ground state is denoted by dash line for reference.
通過分析對比施加應力后材料的態密度和能帶結構,我們發現在 ±5%的面內雙軸應力作用下,材料能隙產生了一定的變化.如圖4所示,在—5%的收縮應力作用下,NaFeF3的能隙約為2.6 eV,而隨著應力逐漸從壓縮向拉伸轉變,材料能隙呈現出單調遞減的變化趨勢,在 +5%的拉伸應力下能隙減小至不足2.4 eV.從圖4(a)—(e)不難看出,材料導帶底隨應力增大而逐漸下移,從而導致了能隙的減小.除此之外,能帶基本結構并未發生顯著改變,價帶頂與導帶底均處于Gamma點,表明在雙軸應力作用下材料為直接帶隙半導體.
3.4 氧元素置換對材料性質的影響
在鈣鈦礦化合物的磁性研究中,離子置換摻雜是一種常用的磁性調控手段[24,25].考慮到Fe的多價態性,Fe3+價態化合物在自然界中相對較多穩定存在,加之O2-離子半徑約為137 pm與F-離子半徑132 pm十分接近,理論上O替換F具有一定的現實可操作性.因此,我們針對O置換F展開了系統研究.
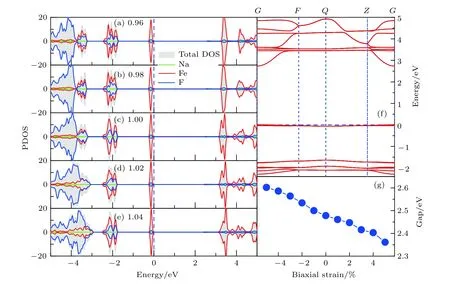
圖4 (a)-(e)面內雙軸應力作用下NaFeF3塊材的態密度分布; (f)NaFeF3塊材能帶結構圖; (g)NaFeF3塊材帶隙隨應力的變化Fig.4.(a)—(e)The pDOS of NaFeF3 with different biaxial strain; (f)the representative band structure of NaFeF3 with —5% in-plane strain; (g)the biaxial strain dependent band gap diagram.
首先,對晶胞中12個F位中的一個進行O離子置換研究,相應的陰離子摻雜濃度約為8.3%.先從能量角度找出最優替換F位,并對替換后的材料磁性進行驗算.由于F八面體結構的特殊性,我們分別對八面體頂點以及FeF2面內的F位進行了置換分析.計算結果表明,面內置換后體系能量要比頂點F位上O離子置換低14 meV,說明O離子更傾向于占據FeF2面內的F位.而無論是頂點還是面內F位的O離子置換,材料G-AFM磁基態始終比亞穩態磁序低100 meV以上,說明8.3%濃度氧離子摻雜對于材料的磁結構未產生顯著影響,G-AFM磁基態非常穩定.
濃度為8.3%的陰離子置換后材料的態密度分布如圖5所示.材料的能帶發生了明顯變化,由于氧離子的摻雜導致導帶底明顯下移,能隙銳減至0.3 eV左右.費米面附近電子態主要源于與O較近的Fe離子的貢獻,Fe的3d和陰離子2p軌道雜化依然較為明顯.F位的O離子置換導致個別Fe離子價態升高,原本占據態的3d軌道穿過費米面變成導帶底.此外,陰離子置換改變了八面體中Fe離子周圍的配位場,使得不同Fe位上的電子態分布也發生了顯著變化,原本對稱分布的自旋反平行的3d電子態,出現了相對位移,如圖5所示.值得注意的是,上述態密度分布變化導致材料價帶頂出現了高自旋極化率的電子態,即所謂雙極化反鐵磁半導體,該類材料在自旋器件設計及自旋電子學研究領域具有巨大的應用價值和研究意義[26].
由于O離子的不對稱摻雜導致八面體中Fe—F和Fe—O鍵長明顯分化,材料結構由Pnma對稱性空間群變為P1空間群.8.3%濃度的O摻雜導致材料沿[001]方向出現FeF2/FeF1.5O0.5/FeF2面交替堆疊,而在FeF1.5O0.5面內形成Fe-O-Fe-FFe-O-Fe的一維鏈.在摻雜面內與O相對接近的Fe進一步被氧化成Fe3+離子,而Fe-O-Fe鏈上另一端的Fe以及近鄰FeF2層內Fe的價態均未受影響,從而呈現出Fe的電荷有序性分布.這一點從計算獲得的Fe離子磁矩上也有所體現: 除與摻雜O成鍵較強的Fe離子上磁矩顯著增大至4.3μB外,該磁矩大小與氧化后的Fe3+離子的3d5高自旋構型相符,其余Fe離子的磁矩仍維持在3.7μB—3.8μB之間.由于材料G型反鐵磁磁序沒有改變,而晶胞中單個Fe離子的價態和磁矩發生了變化,因此材料呈現出亞鐵磁性,并產生0.6μB/u.c.的凈磁矩.
此外,氧離子摻雜導致結構對稱性降低.優化后的結構中,Fe-O-Fe-F-Fe鏈上的Fe—O鍵作用強弱差異導致Fe—O鍵長明顯分化,兩者鍵長差別大于0.1 ?,形成局部偶極矩.通過Berry phase方法計算所得的極化強度約為15 μC/cm2,該數值與點電荷模型估算獲得的極化強度13 μC/cm2相近.隨著FeF2面內方向上電場的調控,Fe-O-Fe鏈中氧離子將在高對稱位置的附近隨外場偏移,從而影響到鏈兩端Fe離子的價態.由于G-AFM的近鄰磁性離子自旋反平行排列的特點,勢必會導致每個原胞內的凈磁矩出現反轉,實現電場對材料磁性的調節.當然實際材料制備過程中,O離子的分布未必如理論模型中這般均勻,因此實際材料中可能會產生局部極化區域,宏觀上表現出弛豫形多鐵現象.
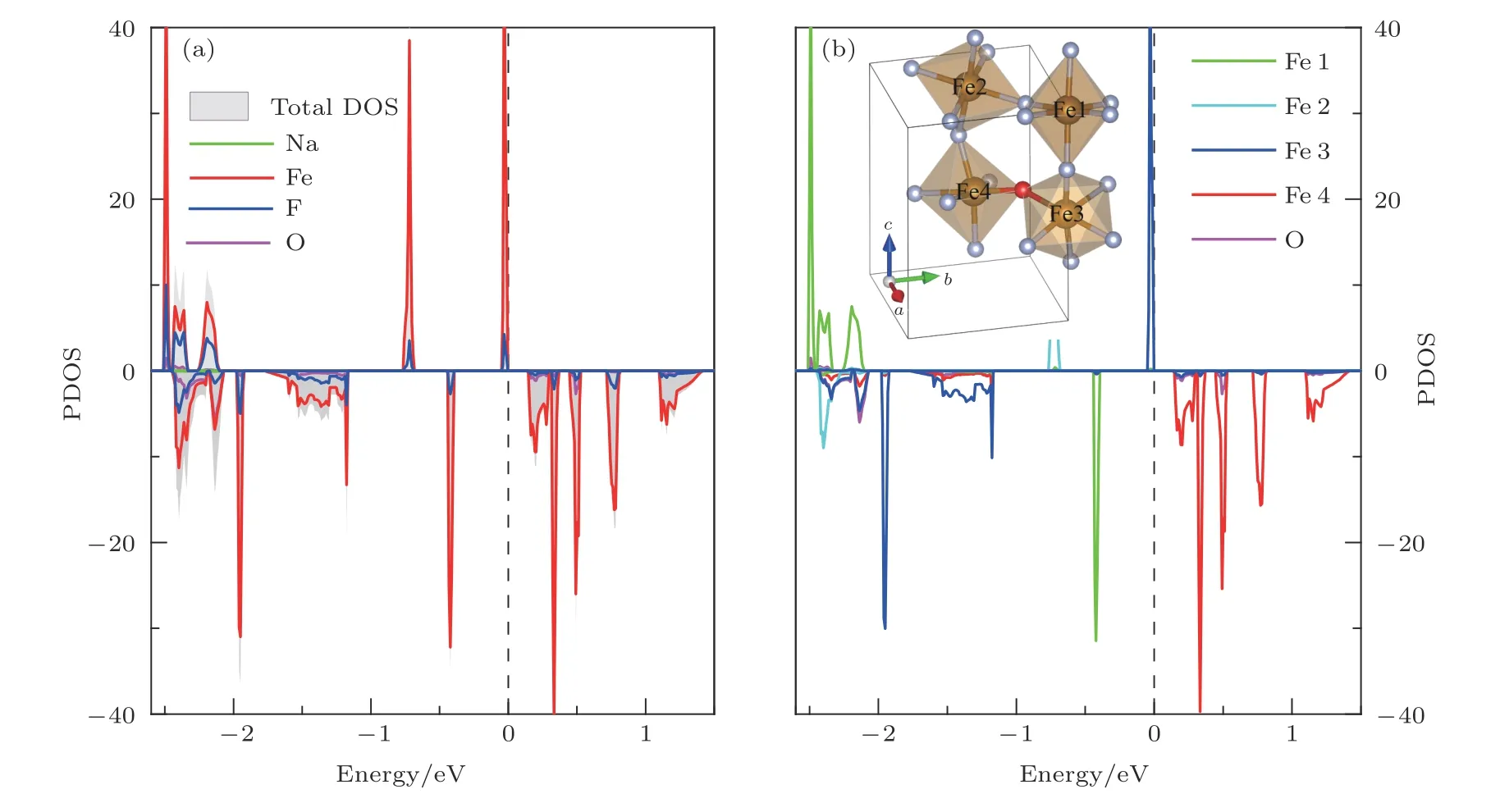
圖5 (a)低濃度氧摻雜時,費米面附近NaFeF3-xOx (x=0.25)的原子分波態密度圖; (b)原胞中不同B位Fe離子的分波態密度,其中插圖為氧離子摻雜后的結構示意圖,為了清晰直觀地表述,圖中僅給出Fe-F(O)八面體結構Fig.5.(a)The pDOS of NaFeF3-x Ox (x=0.25)near Fermi level; (b)the PDOS of different B-site iron ions,which are specified in the inset,Na atoms are omitted for clarify.
接下來,我們將摻雜濃度進一步提高.計算發現NaFeF2O材料中,面內F位上的O離子置換導致體系能量相對于頂點和部分面內頂點F位替換后的體系能量分別低530 meV和12 meV,說明33%的高濃度摻雜情況下,O離子依然傾向于替換FeF2面內的F位,并與近鄰Fe離子形成zigzag形Fe-O-Fe鏈,優化后的晶格結構對稱性屬于Pmc21空間群,通過群論分析不難發現,該對稱群仍屬于極化點群.由于結構中離子位置的非中心對稱性,材料依然可以呈現出一定的鐵電極化強度.但是由于氧摻雜濃度的顯著增多,材料中Fe均被氧化為 +3價,平均磁矩均為4.3μB,在G-AFM磁基態下凈磁矩為零.
NaFeF2O的電荷密度分布如圖6所示.與氧摻雜濃度為8.3%時能隙的急劇減小不同,此時材料的能隙與未參雜塊材能隙相似,約為2.7 eV,處于理想的可見光吸收區.費米面以下直至—2 eV能量范圍內態密度分布主要源自Fe,F和O的電子態,該區域存在著較強的3d和2p軌道雜化.磁性能量對比計算表明,NaFeF2O的磁基態依然是G型反鐵磁序,該反鐵磁序與Fe離子外層3d5電子組態密切相關.與G-AFM的NaMnF3中Mn離子的近鄰磁交換作用相類似[23],近鄰Fe離子半占據的3d軌道通過中間陰離子2p軌道實現反鐵磁超交換.考慮到O2-離子與F-離子半徑相近,陰離子置換并未嚴重影響到材料八面體構型,不同方向上近鄰磁性離子的自旋仍然保持反平行排列,因此G-AFM磁序沒有受到高濃度陰離子置換的影響.此外,費米面以下能帶的顯著展寬與弱氧化性元素置換有助于材料電子態退局域化的趨勢相符.我們還分別嘗試驗算了SrTiO3和LaAlO3襯底應力對材料磁性的影響,結果表明NaFeF2O的磁性仍然沒有出現明顯改變,可見NaFeF3材料的G-AFM磁序不僅不會受到應力作用的影響,對于陰離子摻雜也并不敏感,甚至上述兩種因素共同作用也無法改變材料的反鐵磁基態特性.
4 結 論
至此,我們系統地研究了鈣鈦礦型氟化物NaFeF3的磁電以及結構性質,并通過襯底應力和陰離子摻雜置換的手段,嘗試對其進行了改性調控.計算表明,NaFeF3的G型反鐵磁基態非常穩定,±5%以內的襯底應力以及氧摻雜調控均無法改變該磁序.但是陰離子摻雜卻可以有效改變材料的能帶分布及其能隙大小.當8.3%低濃度氧摻雜時,摻雜面內會出現Fe2+/Fe3+的電荷序,從而產生凈磁矩,同時伴隨著氧離子非對稱偏移導致的局域偶極矩.該偶極矩會受到外電場調控產生極化翻轉,進而影響到Fe離子的電荷序,最終改變凈磁矩的取向.當摻雜濃度提升至NaFeF2O時,材料內Fe離子電荷序消失,凈磁矩也隨之消失.此時材料能隙處于2.7 eV附近,處于理想的可見光吸收波段,在光伏材料設計和光催化應用領域具有較大的研究價值.上述研究結果有助于促進NaFeF3以及與之相類似的鈣鈦礦型氟化物材料的物性研究,同時也為氟化物材料的改性和多功能應用材料的設計研發提供參考.
猜你喜歡
小獼猴智力畫刊(2023年4期)2023-04-23 08:49:58
哲學評論(2021年2期)2021-08-22 01:53:34
中華詩詞(2019年7期)2019-11-25 01:43:04
模具制造(2019年3期)2019-06-06 02:10:54
中學生數理化·高一版(2018年1期)2018-02-10 05:20:03
影視與戲劇評論(2016年0期)2016-11-23 05:26:01
七彩語文·寫字與書法(2016年7期)2016-07-28 21:40:22
七彩語文·寫字與書法(2016年6期)2016-07-15 19:36:34
人間(2015年21期)2015-03-11 15:23:21
現代企業(2015年9期)2015-02-28 18:56:50
