乙酰唑胺關鍵中間體的制備和反應監控
2019-10-15 07:38:50李世文張芳芳
山東化工 2019年18期
關鍵詞:振動
劉 杰,李世文,張芳芳
(河南中帥醫藥科技股份有限公司,河南 鄭州 450000)
乙酰唑胺由Wilier等首先合成,為弱效利尿劑,也曾一度用于心源性水腫和青光眼的治療。有文獻報道了其傳統制法[1]。近年來的研究發現,乙酰唑胺對碳酸酐酶有廣譜抑制作用,可改善生化代謝紊亂,目前臨床應用已拓寬到催眠呼吸暫停綜合癥、老年性癡呆、角膜營養不良水腫、頑固性呃逆等疾病的治療。此外FDA也批準本品用于急性高山病的預防性治療,在急速達到一定海拔高度之前和在此期間,每8~12h口服乙酰唑胺250mg(或每天口服控釋膠囊500mg),可使急性高山病的一些嚴重癥狀明顯減少或者不發生[2-3]。對于乙酰唑胺用于高山病預防性新治療的臨床發現,我們重點研究乙酰唑胺關鍵中間體氯氧化物的制備和反應過程監控,并尋找一種穩定化的工業化生產監控方法。
關鍵中間體氯氧化物(2-乙酰胺基-5-氯磺酰基-1,3,4-噻二唑)的制備是由巰基氧化為磺酰氯基團的過程,大多是采用在酸的水溶液中通入過量的氧化劑氯氣,在低溫條件下將硫醇氧化為磺酰氯,此方法比較傳統,且一直沿用至今,在此不再累述。一直以來,對于氯氧化反應的重點監控是一個空白,一般僅僅通過現象來觀察,具體為描述為:待反應液為黃綠色,反應瓶內固體為白色,且尾氣吸收處的氣體均勻導出時,停止反應[4-8]。顯而易見,此種方式是不科學且不嚴謹的,鑒于以上空白,我們擬采用一種較為科學和有效的方法對氯氧化反應進行監控和跟蹤。
本研究通過高效液相色譜儀(HPLC)對氯氧化反應過程進行監控,并制備得到較優品質的乙酰唑胺,實驗結果表明,采用高效液相色譜儀(HPLC)的監控方法是有效的,且能滿足工業化的需求。
1 實驗部分
1.1 主要儀器與試劑
1.1.1 反應儀器
JJ-1電動攪拌器,低溫恒溫反應浴。
1.1.2 檢驗儀器
高效液相色譜儀,用十八烷基硅烷鍵合硅膠為填充劑的色譜柱。
1.1.3 檢測條件
波長:265nm;柱溫:30℃;流速:1.0mL/min;運行時間:45min。
1.1.4 實驗試劑:
冰醋酸,氨水,硫酸,乙腈,水。
1.1.5 流動相
以0.43%無水醋酸鈉溶液-甲醇-乙腈(95∶2∶3,用冰醋酸調節pH值至4.0士0.05)。
1.2 反應機理
1.2.1 乙酰唑胺合成路線圖
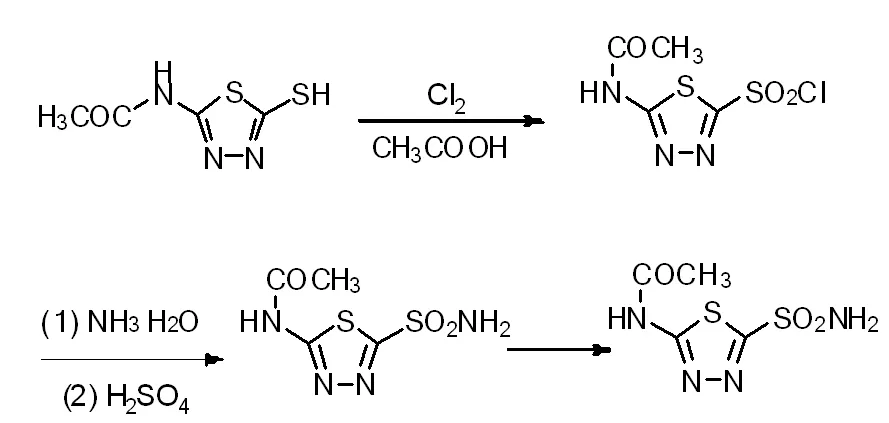
圖1 乙酰唑胺合成路線圖
1.2.2 磺酰氯基團液相監控原理
磺酰氯本身不穩定,在實際檢測中容易變質,導致檢測結果不準或失效,因此對磺酰氯的檢測應先對其進行保護或者取代。本實驗采用的方法為磺酰氯的氨基保護,進而檢測和監控反應的進程,已達到監控的目的。

圖2 磺酰氯基團液相監控原理圖
1.3 實驗過程
1.3.1 氯氧化物(2-乙酰胺基-5-氯磺酰基-1,3,4-噻二唑)的制備
稱量10g的2-乙酰胺基-5-巰基-1,3,4-噻二唑投入到35mL冰醋酸和65mL水的混合體系中,攪拌降溫,5℃下通入氯氣,取樣監控反應。
樣品處理:取樣得到適量的反應液,向反應液中加入氨水,當體系成堿性時,停止加入氨水,攪拌5min,然后加入鹽酸,當體系中析出較多固體時,停止加入鹽酸,攪拌5~10min,抽濾至干,高效液相色譜儀進樣檢測。
體系中的氯氧化反應完全結束后,攪拌保溫1h,將反應體系直接抽濾,水洗滌,60℃干燥,得到氯氧化物(2-乙酰胺基-5-氯磺酰基-1,3,4-噻二唑)。純度97%以上。
1.3.2 乙酰唑胺粗品的制備
將得到的氯氧化物(2-乙酰胺基-5-氯磺酰基-1,3,4-噻二唑)加入到65mL氨水中進行反應,反應完全后,滴加硫酸進行中和,體系達到中性后,停止滴加硫酸,抽濾水洗,60℃真空干燥得到乙酰唑胺粗品。純度98%以上。
1.3.3 乙酰唑胺的精制
將乙酰唑胺粗品、水、焦亞硫酸鈉依次加入到反應瓶中,升溫至回流,降溫析晶,抽濾并水洗,50℃真空干燥,得到乙酰唑胺。純度99%以上。
2 反應監控HPLC圖
2.1 氯氧化物的反應監控液相圖

圖3 反應監控液相圖
注:數據處理方法:面積歸一化法
2.2 乙酰唑胺結構表征
我們將合成得到的乙酰唑胺樣品進行檢測和分析,經UV、IR、1H NMR、13C NMR、HR-MS譜圖分析可知,產品為乙酰唑胺,具體分析如下:
(1)高分辨質譜分析表明樣品的準分子離子峰[M+H]+為222.9953,可推斷其分子量為221.9880,與乙酰唑胺的理論精確分子量221.9881相比,誤差小于3ppm,元素匹配結果表明其元素組成為C4H6N4O3S2,與乙酰唑胺的結構一致,元素分析結果表明其元素組成正確。
(2)IR:3301.90cm-1:磺酰胺N-H的伸縮振動,3182.26 cm-1:乙酰胺基N-H的伸縮振動,1551.81cm-1:N-H的彎曲振動;1320.54cm-1:磺酰胺SO2的不對稱伸縮振動,1176.61cm-1:磺酰胺SO2的對稱伸縮振動,說明分子中含有磺酰胺基和乙酰胺基。1H NMR上δ13.013(1H,br,s)、8.333ppm(2H,br,s)的活潑氫,證明分子中存在NH和NH2。
(3)IR:2906.42,2783.91cm-1:乙酰甲基C-H的伸縮振動,1434.89cm-1:乙酰甲基C-H的彎曲振動,1680.38cm-1:酰胺羰基C=O 的伸縮振動,說明分子中含有乙酰胺基。1H NMR上δ2.255(3H,s)ppm的甲基質子和13C NMR譜上δ22.326(伯C)ppm、13C NMR譜上δ169.376(季C)ppm的乙酰羰基碳,進一步證明了它的存在。
(4)IR:1551.81cm-1:噻二唑C=N 的伸縮振動,證明分子中有C=N。13C NMR譜上δ164.263(季C)、δ161.085(季C)ppm進一步證明了它的存在。
綜上所述,分子中含有磺酰胺基、乙酰胺基和噻二唑等基團,與乙酰唑胺的結構相符,樣品的UV、IR、11H NMR、13C NMR、HR-MS譜圖可由乙酰唑胺的結構給以合理的解釋,證明樣品結構與乙酰唑胺結構一致。
3 結果與討論
3.1 氯氧化過程中氯氣用量的選擇
氯氣作為氯氧化反應過程中重要的反應物料,需對其用量做以研究,本實驗組對氯氣用量做以考察,實驗結果匯總如下。

表1 氯氣用量對氯氧化反應的影響結果匯總
由氯氣用量對比試驗可知,隨著氯氣用量的增加,產品的收率和純度都隨之增加,在氯氣用量達到1.9~2.2倍時,產物(粗品)的純度已達到較好的水平,收率尚可,因此我們可暫選此用量范圍作為我們后續實驗的基礎,選擇氯氣用量為AMTD用量的1.9~2.2倍。
3.2 氯氧化過程中反應溫度的選擇
反應溫度作為一個重要的工藝參數,我們要對其進行考察,擬采用不同反應溫度進行反應,以觀察和比較不同反應溫度對氯氧化反應的影響,試驗數據如下:
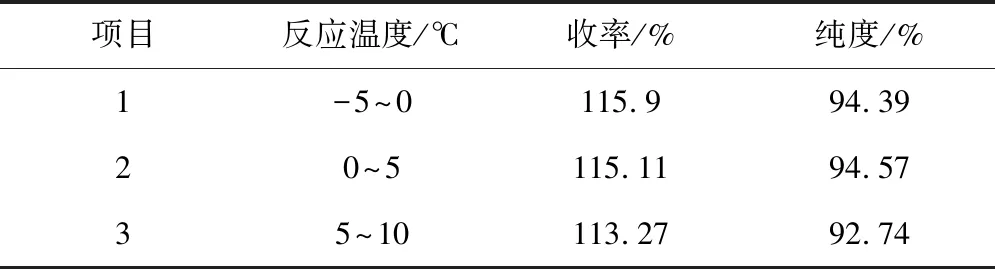
表2 反應溫度對氯氧化反應的影響結果匯總
由以上試驗可知,在不同的溫度條件下進行氯氧化反應,3批實驗的收率差別不大,但純度差異較大,其中5~10℃條件下,純度較低,不采用此溫度條件,優先采用0~5℃為反應溫度(氯氧化反應)。
3.3 氯氧化過程中的反應時間
通過反應監控來觀察反應進程,判斷較優反應時間,使反應時間達到最優化,以利用整個反應過程,試驗結果如下:

表3 反應時間對氯氧化反應的影響結果匯總
通過反應監控可知,氯氣量達到時,反應已進行了大半,后續反應仍在進行,但是反應較慢,鑒于此種情況,我們暫定反應時間為60min。
4 結論
在乙酰唑胺的合成中,我們重點研究了氯氧化反應過程,氯氧化物的反應增加了HPLC監控的方法,使我們能夠更加清晰和準確的掌控反應進程,進而得到質量更優的產品。經UV、IR、1H NMR、13C NMR、HR-MS譜圖分析,我們得到的產品為目標產物乙酰唑胺。
猜你喜歡
科學大眾(2023年17期)2023-10-26 07:39:14
大電機技術(2022年5期)2022-11-17 08:12:48
天天愛科學(2020年6期)2020-09-10 07:22:44
瘋狂英語·新讀寫(2020年3期)2020-06-06 09:05:56
數學物理學報(2018年4期)2018-09-14 03:40:58
數學物理學報(2017年6期)2018-01-22 02:26:40
船海工程(2015年4期)2016-01-05 15:53:26
噪聲與振動控制(2015年4期)2015-01-01 07:08:44
計算物理(2014年2期)2014-03-11 17:01:44
鄭州大學學報(理學版)(2014年3期)2014-03-01 04:21:00
