液相色譜-串聯質譜法同時測定丹參中139種農藥殘留
2019-09-10 07:22:44李雯婷苗水陳銘周恒蘭嵐毛秀紅季申
世界中醫藥 2019年4期
關鍵詞:檢測
李雯婷 苗水 陳銘 周恒 蘭嵐 毛秀紅 季申
摘要?目的:采用液相色譜-串聯質譜法建立丹參中農藥多殘留的測定方法。方法:樣品經改良的QuEChERS方法提取,采用分散固相萃取(d-SPE)二次凈化法凈化,并采用液相色譜-串聯質譜技術在多反應監測(MRM)模式下測定,內標法定量。結果:丹參中139種農藥在0.005~0.2 mg/L范圍內線性關系均良好;在10、50和100 μg/kg添加水平下,除了噻菌靈回收率偏低(67.2~67.9)外,其余農藥的平均回收率為70.2%~126.1%,相對標準偏差(RSD)為1.2%~19.5%,139種農藥的檢測限為0.005~0.05 mg/kg。結論:該方法樣品前處理簡單快速,靈敏度、準確度和精密度均符合農藥多殘留檢測技術的要求,適用于丹參中139種農藥殘留的快速篩查測定。
關鍵詞?液相色譜-串聯質譜法;農藥殘留;QuEChERS法;丹參;分散固相萃取;固相萃取;鹽析劑;勻漿法
Simultaneous Determination of 139 Pesticide Residues in Salvia miltiorrhiza by LC-MS/MS
Li Wenting,Miao Shui,Chen Ming,Zhou Heng,Lan Lan,Mao Xiuhong,Ji Shen
(Shanghai Institute for Food and Drug Control,Shanghai 201203,China)
Abstract?Objective:To study the multi-residues detection methods for Salvia miltiorrhiza by LC-MS/MS.Methods:A method using a modified QuEChERS and d-SPE clean-up followed by LC-MS/MS has been established for the quantitative determination of the residues.The extract was determined by LC-MS/MS in multi-reaction monitoring(MRM)mode,and internal standard method was applied to quantify the pesticides.Results:All the 139 pesticides showed good linearity in the range of 0.005~0.2 mg/L.The average recoveries of the pesticides at the spiked levels of 10,50,100 μg/kg ranged from 70.2~126.1% with the RSDs of 1.2~19.5%,except for Thiabendazole(67.2~67.9%).The limits of detection(LODs)of 139 pesticides were 0.005~0.05 mg/kg.The results demonstrated that this method was simple,rapid and suitable for the determination of 139 pesticide residues in Salvia miltiorrhiza.Conclusion:The analytical sensitivity and accuracy can meet the requirements of multiple pesticide residue analysis.
Key Words?LC-MS/MS; Pesticide residues; QuEChERS; Salvia miltiorrhiza; dSPE; SPE; Salting-out agent; Homogenate
中圖分類號:R284.1文獻標識碼:Adoi:10.3969/j.issn.1673-7202.2019.04.001
丹參(Salviae Miltiorrhizae Radix et Rhizoma)為唇形科植物丹參(Salvia miltiorrhiza Bge.)的干燥根和根莖,具有活血祛瘀,通經止痛,清心除煩,涼血消癰的功效[1]。臨床應用范圍廣泛,屬于大宗常用中藥材。在丹參大規模人工種植情況下,農藥的使用是保障丹參產量與質量的重要手段。但隨著農藥的廣泛使用,丹參農藥殘留問題日趨嚴重,直接影響了人民用藥的安全性和有效性,也是制約中藥走向國際市場的瓶頸。因此,建立簡便、高效的丹參中農藥多殘留檢測方法,對保證用藥安全及推進中藥現代化、國際化具有重要的意義。
農藥多殘留分析亟需解決的主要問題之一是去除基質干擾對于殘留農藥定量準確度、靈敏度及儀器污染的影響。因此,前處理中凈化方法的選擇和優化是農藥多殘留檢測的關鍵。目前,常用的前處理技術主要包括固相分散萃取[2-4]、固相微萃取[5]、凝膠滲透色譜[6]以及QuEChERS[7-8]等。而QuEChERS法以其快速、簡便、高效、安全等特點,已廣泛應用于中藥材農藥多殘留分析,是當前國內外實驗室普遍采用的前處理方法。
儀器檢測方法主要有氣相色譜法(GC)[9-11]、氣相色譜-質譜法(GC-MS)[12-13]、氣相色譜-串聯質譜法(GC-MS/MS)[14-17]、液相色譜-質譜法(LC-MS)[18]以及液相色譜-串聯質譜法(LC-MS/MS)[19-22]等。常用的色譜法由于檢測器的局限,多農藥篩查能力較弱;且由于丹參基質的干擾,無法通過保留時間定性定量,易產生假陽性和假陰性的結果。GC-MS和LC-MS被廣泛應用于農藥多殘留檢測中,但2種檢測方法對于幾百種農藥的多殘留檢測需建立多個檢測方法,樣品需反復測定,耗時耗力;且在選擇離子檢測(SIM)模式下采集的質譜信息少,選擇性較差,結果存在較大的不確定性。液相色譜-三重四極桿串聯質譜通過其高通量的離子傳輸性能,在高選擇性的多反應監測(MRM)模式下可實現一次進樣同時分析百余種化合物,且基質干擾少,能有效減少和避免假陽性和假陰性的現象,是中藥材農藥多殘留檢測的必然選擇。丹參中存在大量的脂溶性色素和水溶性酚酸等,對農藥殘留測定干擾大。本文以改良的QuEChERS法提取,采用分散固相萃取(d-SPE)二次凈化法凈化,并采用LC-MS/MS在MRM模式下進行測定,內標法定量,快速準確地測定了丹參中139種農藥殘留。該方法具有操作簡便、靈敏度高、應用廣泛等特點,對丹參藥材的安全監測有重要的意義。
1?儀器與試藥
1.1?儀器
Agilent 1290超高效液相色譜儀(美國Agilent公司),AB SCIEX 5500三重四極桿質譜儀(美國AB SCIEX公司),旋轉蒸發儀(瑞士BüCHI公司),N-EVAPTM 112型氮吹儀(美國Organomation公司),勻漿機(上海Fluko公司),IKA KS260C振蕩器(德國IKA公司),VisiprepTM DL固相萃取裝置(美國Supelco公司),IKA MS 2渦旋混合器(德國IKA公司),UV2550(日本島津公司)
1.2?試劑與材料
139種農藥標準品(Dr.Ehrenstorfer GmbHr公司或Chem Service公司);乙腈、甲醇、丙酮、乙酸均為色譜純;無水醋酸鈉,無水硫酸鎂為分析純;C18固相萃取柱(400 mg/6 mL)、石墨化碳:乙二胺-N-丙基硅烷混合型固相萃取柱(PC/PSA-SPE)(400 mg:400 mg/6 mL);吸附劑:N-丙基乙二胺(PSA)、弗羅里硅土(Florisl)、石墨化炭黑(GCB)、十八烷基鍵合硅膠(C18)、硅膠(Silica)、Chlorofiltr均購于天津博納艾杰爾科技有限公司;丹參藥材均購于上海華宇藥業。
1.3?分析樣品
實驗用丹參樣品分別來源于上海華宇藥業有限公司、上海養和堂中藥飲片有限公司、上海中西制藥有限公司共9批,依次編號為1~9。
2?方法與結果
2.1?LC-MS/MS色譜質譜條件
色譜條件:色譜柱:Agilent SB-C18(3.5 μm,2.1×150 mm);流動相:A相,0.1%甲酸+10 mmol/L甲酸鈉水溶液;B相,乙腈。梯度洗脫程序:0~15.00 min,75% A~10% A;15.00~20.00 min,10% A~10% A;20.00~28.00 min,75% A~25% A。流速:0.40 mL/min;進樣量:2.0 μL。
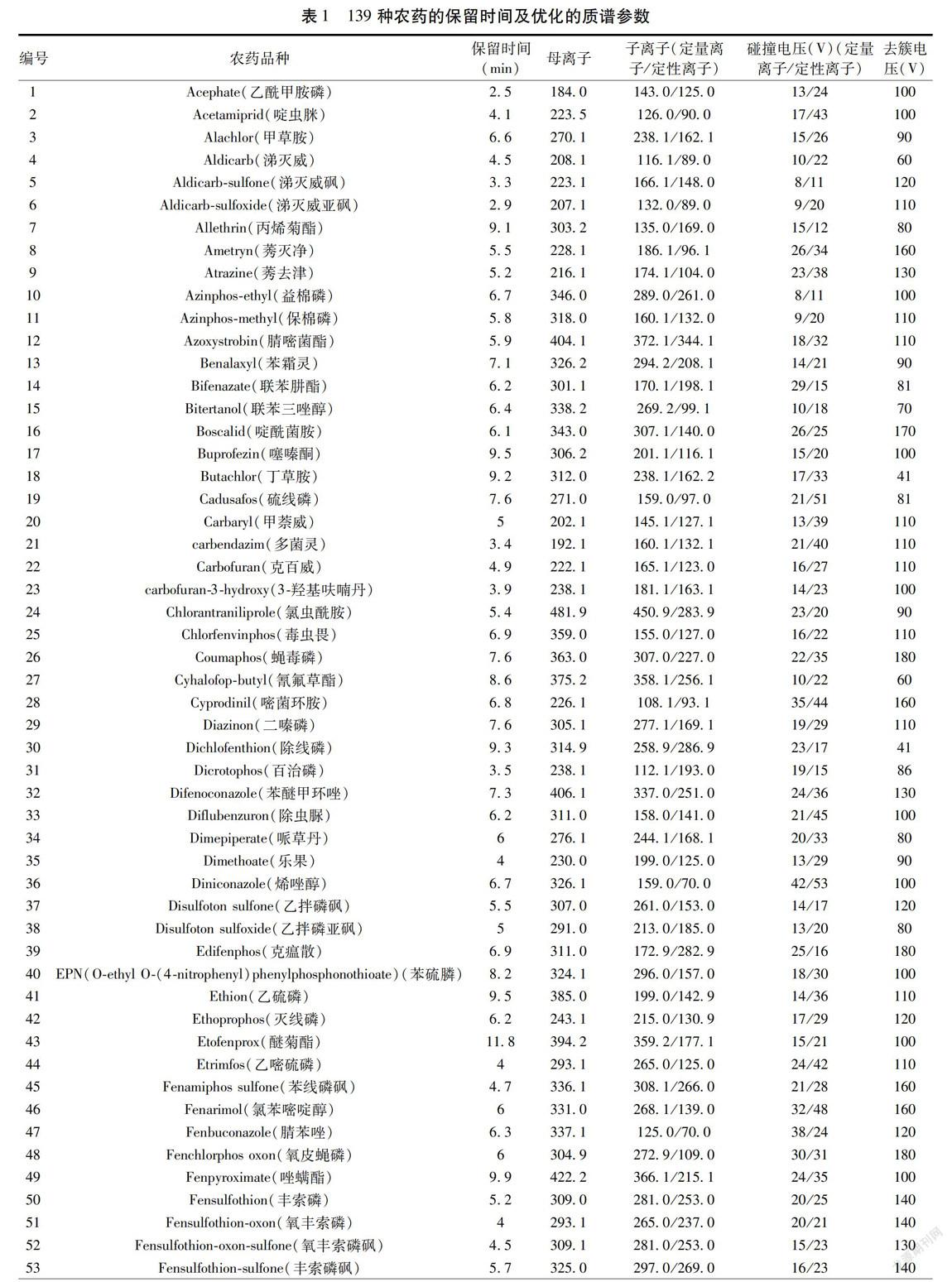
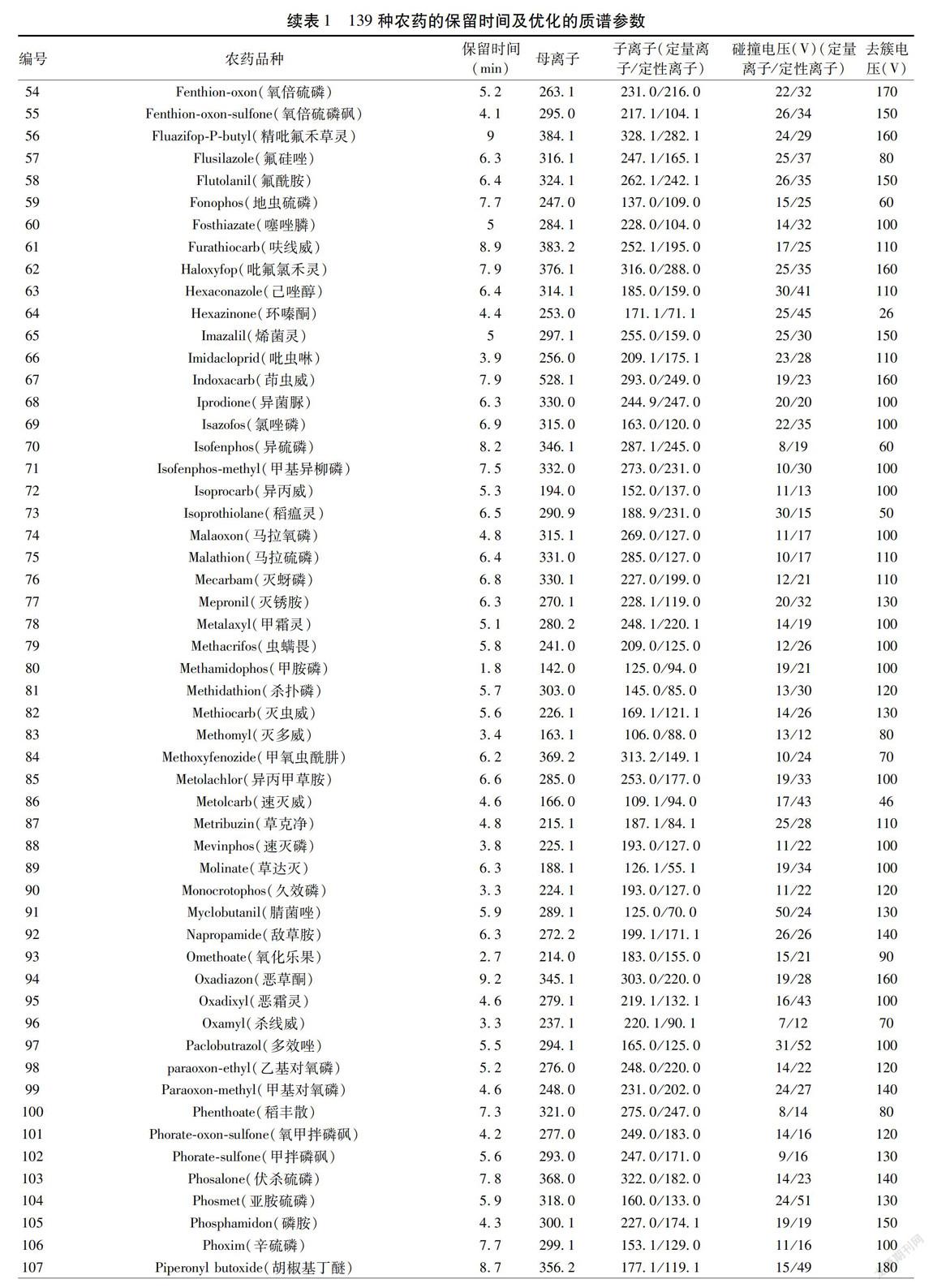
質譜條件:離子源:電噴霧離子源(ESI);掃描方式:正離子掃描;檢測方式:多反應監測(MRM)模式;電噴霧電壓:5 500 V;霧化氣壓力:50.0 psi;輔助氣壓力:50.0 psi;氣簾氣壓力:20.0 psi;碰撞氣壓力:7.0 psi;離子源溫度:500 ℃;掃描時間:50 ms;碰撞室入口電壓:10 V;碰撞室出口電壓:12 V;監測離子對及其相關質譜參數見表1。按照上述LC-MS/MS條件所得139種農藥MRM圖譜見圖1。
2.2?標準品溶液的配制
標準品儲備溶液的制備:精密稱取各農藥對照品適量,根據各農藥溶解性加乙腈或甲苯分別制成每1 mL中含1 000 μg的溶液,即得,貯存在4 ℃冰箱中備用。
內標儲備溶液的制備:精密稱取氘代莠去津(Atrazine-d5)標準品適量,加乙腈溶解并制成每1 mL含1 000 μg的溶液,即得。
混合標準品溶液的制備:精密量取上述標準品儲備液適量,用含0.05%醋酸的乙腈分別制成每1 mL含100 μg和 1 000 μg的2種溶液,即得。
內標溶液的制備:精密量取內標儲備溶液適量,加乙腈制成每1 mL含6 μg的溶液,即得。
基質混合標準品工作溶液的制備:取丹參空白基質樣品3 g,一式6份,同供試品溶液的制備方法處理至“吸取上清液0.5 mL”,置氮吹儀上于40 ℃水浴濃縮至約0.2 mL,分別加入混合標準品溶液(100 μg/L)25 μL、50 μL,混合標準品溶液(1 000 μg/L)25 μL、50 μL、100 μL、200 μL,加乙腈定容至0.5 mL,渦旋混勻,用微孔濾膜濾過(0.22 μm),取續濾液,即得系列基質混合標準品工作溶液。
2.3?供試品制備方法
提取:取供試品,粉碎成粉末(過三號篩),取約3 g(精確至0.01 g),置50 mL聚苯乙烯具塞離心管中,加入1%冰醋酸溶液15 mL,渦旋使藥粉充分浸潤,放置30 min,精密加入乙腈15 mL與內標溶液100 μL,渦旋使混勻,置振蕩器上劇烈振蕩(500次/min)5 min,加入無水硫酸鎂與無水醋酸鈉的混合粉末(4∶1)7.5 g,立即搖散,再置振蕩器上劇烈震蕩(500次/min)3 min,于冰浴中冷卻10 min,離心(4 000 r/min)5 min,待凈化。
凈化:精密移取上清液9 mL置已預先裝有凈化材料的分散固相萃取凈化管(弗羅里硅土300 mg,十八烷基鍵合硅膠300 mg,N-丙基乙二胺600 mg,石墨化炭黑150 mg,無水硫酸鎂900 mg)中,渦旋使充分混勻,再置振蕩器上劇烈震蕩(500次/min)5 min使凈化完全,離心(4 000 r/min)5 min,精密吸取上清液5 mL,置氮吹儀上于40 ℃水浴濃縮至約0.4 mL,加乙腈定容至1 mL,渦旋使混勻,加入N-丙基乙二胺50 mg、無水硫酸鎂150 mg,渦旋60 s使凈化完全,離心(4 000 r/min)5 min,取上清液0.5 mL,渦旋混勻,用微孔濾膜(0.22 μm)濾過,取續濾液,即得。
2.4?線性范圍和檢出限
按照“2.2”項下的方法配置基質標準溶液,進行測定。以各組分的質量濃度為橫坐標(X),各組分的峰面積與內標峰面積比為縱坐標(Y),進行回歸分析,結果見表2。139種農藥在0.005~0.2 mg/kg線性范圍內線性關系良好,相關系數(r)均≥0.99。采用在空白丹參基質中添加較低濃度水平的混合標準品溶液的方法,確定各農藥在丹參基質中的最低檢出限,本實驗是以實際看到峰的最低濃度作為檢出限,檢出限結果見表2,從表2可得約90%農藥的檢出限可到達0.005 mg/kg,說明方法的靈敏度高。
2.5?回收率和精密度
將混合標準品儲備溶液添加到丹參空白基質中,分別制成質量濃度為10、50、100 μg/kg的加標樣品溶液,以“1.3”所述方法進行前處理,一式6份,計算回收率和相對標準偏差(RSD)。3個加標水平下,除了噻菌靈回收率偏低(67.2~67.9),其余農藥的平均回收率為70.2%~126.1%,RSD為1.2%~19.5%,數據表明該方法具有良好的適用性,回收率滿足農藥殘留檢測要求。
2.6?樣品測定結果
采用所建立的分析方法對9批丹參藥材進行農藥殘留檢測,樣品測定結果見表3。
3?討論
3.1?檢測農藥品種的選擇?根據我國中藥材中農藥的施用情況,并參照國內外農藥殘留監測動向,依據以下原則選擇檢測農藥的品種:國內國際禁限用農藥品種的名單;在我國有關部門登記常用農藥中毒性較大,對人類存在“三致”危險的農藥;我國食品國家標準(GB)中監控的農藥,且毒性較大的農藥;美國、歐洲等各國藥典針對植物藥的農藥殘留監控的品種。根據以上原則篩選的農藥品種再結合液相色譜串聯質譜分析的特點,最終選擇檢測農藥為139種。
3.2?提取方法的優化
3.2.1?提取方式的選擇?選用丹參陽性樣品(檢出多菌靈),分別采用加水浸泡后提取的改良QuEChERS法與直接用乙腈提取的勻漿法提取,實驗發現改良QuEChERS法的藥材溶脹率為勻漿法的3倍左右,且改良QuEChERS法的提取效率明顯高于勻漿法,這是由于中藥材丹參含水量較少,直接使用有機溶劑難以有效提取目標分析物;而加水溶脹后提取溶劑可滲入細胞及組織內部,提高提取效率,故最終采用改良QuEChERS法提取。
3.2.2?提取溶劑的選擇?目前,農藥多殘留檢測常用的提取溶劑有乙酸乙酯、丙酮和乙腈。乙酸乙酯由于與水不互溶,不易滲入植物細胞,不利于農藥萃出。本研究比較了丙酮、乙腈2種提取溶劑,結果顯示(圖2),丙酮提取液的顏色遠深于乙腈,說明提取液中共萃物的雜質較多,對后續凈化與分析過程產生不利影響,且丙酮的沸點較低,易于揮發,提取時易造成提取溶劑的體積變化;而乙腈適合的農藥極性范圍較廣,可以有效萃取丹參中的農藥殘留同時減少糖類、色素與脂肪類成分等共萃物的提取,故而選用乙腈作為提取溶劑。
3.2.3?浸泡時間的考察?本實驗考察了不同浸泡時間對提取效率的影響。選用丹參陽性樣品(檢出多菌靈)加水分別浸泡0.5、1、3 h,然后加乙腈振蕩提取。結果可見,不同浸泡時間對于陽性樣品表現出一致的提取效率,說明浸泡0.5 h即可將殘留農藥提取完全,故最終確定浸泡時間為0.5 h。
3.2.4?鹽析劑的選擇?QuEChERS法中鹽析劑的使用可以促進目標化合物由水相分配至有機相中。本實驗比較了NaCl和NaAc 2種鹽析劑,實驗發現2種鹽析劑對農藥回收率的影響沒有明顯的區別,平均回收率分別為102.7%和98.3%。但在提取液中加入NaAc,可以使提取液形成HAc-NaAc緩沖系統,保證萃取液的pH值穩定在4~5之間,從而減少或防止在堿性或中性環境中不穩定農藥的降解,提高了方法的適用范圍;且采用NaAc作為鹽析劑的提取液顏色明顯淺于NaCl(圖2),說明其中色素等共提取較少,故最終選擇NaAc作為鹽析劑。
3.3?凈化條件的優化
3.3.1?凈化方法的選擇
目前,農藥多殘留的常用凈化方法有固相萃取法(SPE)和分散固相萃取法(d-SPE)。本實驗比較了固相萃取法(C18柱/500 mg、GCB/PSA串聯柱/400 mg/400 mg/)、一次分散固相萃取凈化法(Florisl/300 mg、C18/300 mg、PSA/300 mg、GCB/150 mg、無水硫酸鎂/900 mg)、二次分散固相萃取凈化法(①Florisl/300 mg、C18/300 mg、PSA/300 mg、GCB/150 mg、無水硫酸鎂/900 mg②PSA50 mg,無水硫酸鎂150 mg)。通過比較凈化液的顏色深淺來評價凈化效果,實驗發現二次d-SPE凈化法效果最佳,SPE法次之,一次d-SPE凈化法稍差;并且二次d-SPE法較SPE法操作簡便,重復性好,溶劑用量少,耗時短,故本實驗選擇二次d-SPE凈化法作為凈化方法。
3.3.2?凈化材料的優選
3.3.2.1?一次凈化材料的選擇
針對丹參基質主要含有脂溶性色素類、水溶性酚酸類、黃酮類、三萜類和甾醇類等干擾成分。本實驗比較了C18、PSA、Florisl、Silica、GCB和Chlorofiltr等六種凈化材料。其中GCB與Chlorofiltr均可用于除去基質中的色素類雜質,但GCB的凈化效果明顯好于Chlorofiltr,且用量較少,故選擇GCB作為凈化材料;Florisl和Silica均可用于除去羥基類及含雜原子或雜環化合物的極性干擾物,但Florisl的凈化效果較好,故選擇Florisl作為凈化材料;C18可去除丹參基質中的脂類和甾醇類干擾成分;PSA可去除與GCB不同的有機酸和極性色素類干擾成分。故最終優選GCB、C18、PSA及Florisl作為凈化材料,4種凈化材料具有不同的凈化機理,一次凈化,可去除多種干擾成分。
3.3.2.2?二次凈化材料的選擇
針對丹參中色素類干擾成分多的特點,選擇了GCB、PSA和Florisl 3種凈化材料進行考察,實驗發現GCB/15 mg與PSA/50 mg凈化效果相當,Florisl/50 mg稍差;由于GCB對個別含平面及對稱結構的農藥有一定的吸附作用,故最終選擇PSA/50 mg作為二次凈化的凈化材料。
3.3.3?凈化材料配比的考察
3.3.3.1?GCB用量的考察
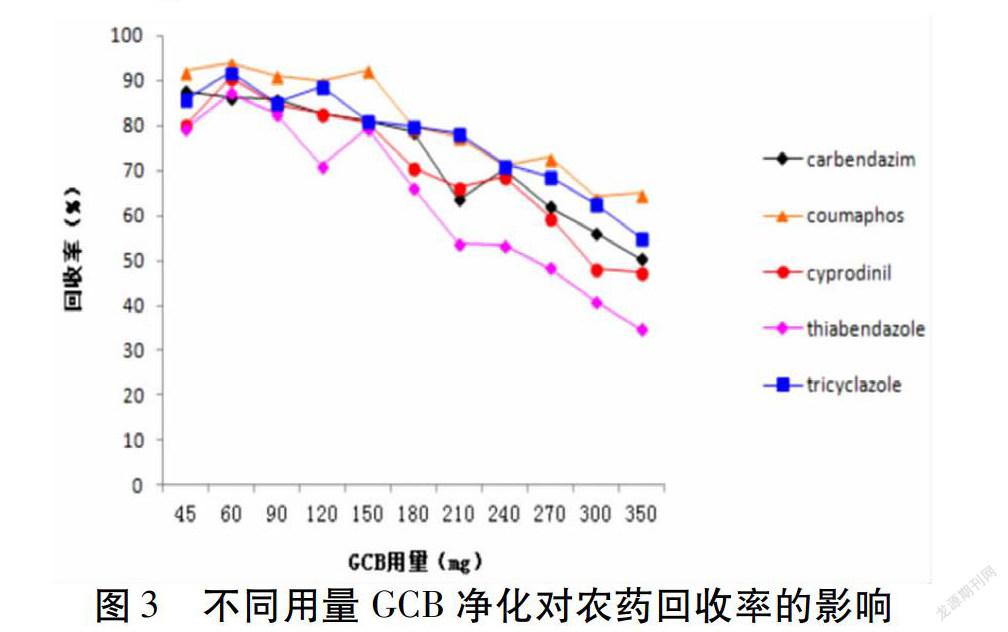
GCB雖然可以有效地去除色素,但由于其表面的正六元環結構會吸附一些平面及對稱結構的農藥(如多菌靈、蠅毒磷、嘧菌環胺、噻菌靈、三環唑等),導致這些組分回收率降低。所以GCB用量要根據基質所含色素干擾成分的多少決定,使其既能起到凈化作用又不降低這類農藥的回收率。本實驗對不同用量的GCB進行了考察,由圖3可見多菌靈、蠅毒磷等5種農藥隨著GCB用量的增加,回收率逐漸減小;當GCB用量為150 mg時,回收率符合要求,所以確定GCB的用量為150 mg。
3.3.3.2?PSA、C18、Florisl用量的考察
在石墨化炭黑用量確定的基礎上,對PSA、C18和Florisl 3種凈化材料的用量進行考察。采用紫外光譜掃描,分別比較了3種凈化材料的用量為300 mg、600 mg、900 mg時的凈化效果,實驗發現隨著Florisl、C18用量的增加,對丹參提取液的凈化效果沒有明顯的提升;而隨著PSA用量的增加,對丹參提取液的凈化效果提升明顯,故確定增加PSA的用量。
考察了PSA的用量對農藥回收率的影響,實驗發現增大PSA用量,雖然凈化效果明顯,但對部分農藥的回收率產生影響,不能滿足檢測要求;而當PSA用量為600 mg時,各農藥的回收率最佳,因此最終確定凈化材料的配比為Florisl/300 mg,C18/300 mg,PSA/600 mg,GCB/150 mg,無水硫酸鎂/900 mg。
3.4?基質效應的評價
基質效應是指基質中共提取干擾物影響目標化合物的離子化,從而使化合物在儀器上的響應發生增強或抑制。因此,需要對目標化合物的基質效應進行評價,并根據評價結果選擇適合的方法對基質效應進行抵消。本試驗通過測定溶劑標準溶液和基質標準溶液,分別建立標準曲線,比較兩條標準曲線斜率的差異,從而判斷基質效應的強弱,計算公式如下:基質效應=[(基質匹配標準曲線的斜率/溶劑標準曲線的斜率)-1]×100%。實驗發現,139種農藥中,僅16種農藥的基質效應不明顯(±30%),其余123種農藥均產生中等和較強的基質抑制效應(<-30%)。由于基質抑制效應明顯,最終采用基質匹配內標標準曲線法對樣品中的農藥殘留進行定量,以抵消基質效應的影響。
3.5?丹參檢測結果分析
9批丹參藥材中有7批檢出1~2種農藥,檢出率為78%;檢出農藥分別為多菌靈、吡蟲啉、啶蟲脒;且有6批藥材均檢出多菌靈,提示多菌靈為丹參種植常用農藥,需加強丹參中多菌靈的監測。
本研究采用改良的QuEChERS提取,針對丹參基質研發的二次分散固相萃取法凈化,建立了液相色譜-串聯質譜同時測定丹參中139種農藥殘留的分析方法。該方法樣品前處理簡單,靈敏度、準確性均能滿足農藥殘留檢測要求,為中藥材丹參中多類別農藥殘留的快速篩查、風險評估等研究提供了一種高效、可靠的分析手段。
參考文獻
[1]國家藥典委員會.中華人民共和國藥典[M].北京:中國醫藥科技出版社,2015:76-77.
[2]苗水,郟征偉,毛秀紅,等.氣相色譜串聯質譜法同時測定黃芪中238種農藥殘留[J].中國藥學雜志,2012,47(4):303-310.
[3]李繼革,施家威,王玉飛.固相萃取-氣相色譜-串聯質譜法測定絞股藍中75種農藥殘留[J].中國衛生檢驗雜志,2017,27(19):2746-2753.
[4]劉運明,李放.固相萃取-氣相色譜同時測定含西洋參保健食品中18種有機氯農藥殘留[J].江蘇預防醫學,2017,28(6):706-707,710.
[5]吳麗蘋,曾憲錄,盧任杰,等.SPME-GC-MS法測定人參中6種有機氮農藥殘留[J].食品工業,2018,39(1):286-289.
[6]何曼莉,謝建軍,陳捷,等.凝膠滲透色譜-固相萃取-氣相色譜法同時測定藥食兩用中藥材中17種有機氯類農藥殘留量[J].食品安全質量檢測學報,2015,6(3):893-901.
[7]梁利鵬,丁輝,余河水,等.QuEChERS法結合氣相色譜-質譜快速測定人參中33種農藥殘留[J].遼寧中醫藥大學學報,2017,19(9):63-67.
[8]王建國,魏玉霞,王芳,等.氣相色譜-三重四級桿串聯質譜法快速檢測中草藥中85種農藥殘留[J].中國衛生檢驗雜志,2017,27(3):324-330.
[9]李妮娜,王舉濤.氣相色譜法對丹參中有機氯農藥殘留的分析研究[J].安徽醫藥,2015,19(4):656-659.
[10]沈廷明,劉知遠,吳仲玉,等.氣相色譜法測定金線蓮中六六六和滴滴涕農藥殘留[J].中草藥,2016,47(22):4082-4084.
[11]陸繼偉,苗水,毛秀紅,等.固相萃取-氣相色譜雙塔雙柱法同時測定中藥材中53種有機磷類農藥殘留量[J].中成藥,2010,32(1):94-99.
[12]劉洪波.氣質聯用測定杭白菊等3種中藥中5種擬除蟲菊酯類農藥殘留[J].浙江農林大學學報,2015,32(1):110-115.
[13]林林,劉廣楨,穆向榮,等.氣相色譜-質譜法測定瓜蔞中的70種農藥殘留[J].中國衛生檢驗雜志,2017,27(9):1217-1221.
[14]苗水,郟征偉,毛秀紅,等.液相色譜-串聯質譜法同時測定穿心蓮中敵克松、甲基托布津、甲萘威和敵百蟲的農藥殘留量[J].藥物分析雜志,2011,31(3):529-533.
[15]田紹瓊,毛秀紅,苗水,等.氣相色譜-串聯質譜法測定人參和黃芪中7種毒殺芬殘留量[J].色譜,2012,30(1):14-20.
[16]程志,張蓉,劉韋華,等.氣相色譜-串聯質譜法快速篩查測定中藥材中144種農藥殘留[J].色譜,2014,32(1):57-68.
[17]王建國,萬麗葵,魏玉霞,等.氣相色譜-串聯質譜同位素內標法測定中草藥中124種農藥及其代謝物殘留[J].分析試驗室,2017,36(2):220-225.
[18]劉銀,龔婧如,毛秀紅,等.高效液相色譜-質譜聯用法測定白芍藥材中23種農藥殘留[J].農藥學學報,2011,13(5):496-502.
[19]林濤,陳興連,姚清華,等.超高效液相色譜-串聯質譜法快速測定金銀花中77種農藥殘留[J].分析試驗室,2018,37(9):1037-1044.
[20]俞建忠,蒼濤,戴芬,等.超高效液相色譜-串聯質譜法測定浙貝母中6種農藥殘留[J].農藥學學報,2018,20(3):370-376.
[21]黃莉莉,李麗莉,羅軼,等.LC-MS/MS法測定桑葉藥材中100種農藥殘留量[J].中國藥房,2016,27(15):2122-2126.
[22]鄭尊濤,龔勇,孫豐收,等.高效液相色譜-串聯質譜法同時測定枸杞中24種農藥殘留[J].農藥,2018,57(11):826-828,839.
猜你喜歡
中國設備工程(2022年12期)2022-07-11 04:33:00
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2020年12期)2021-01-18 06:57:46
中學生數理化·七年級數學人教版(2020年12期)2021-01-18 06:57:46
中學生數理化·七年級數學人教版(2019年9期)2019-11-25 07:34:36
中學生數理化·七年級數學人教版(2019年9期)2019-11-25 07:34:34
中學生數理化·七年級數學人教版(2019年12期)2019-05-21 02:53:50
中學生數理化·七年級數學人教版(2019年12期)2019-05-21 02:53:48
